Buy article online - an online subscription or single-article purchase is required to access this article.
The title compound, C
20H
10Cl
2N
2O
2, is one of the quinacridone derivatives known as red pigments. The molecule has inversion symmetry. The quinacridone molecules are connected by N—H

O hydrogen bonds along the [1
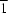
0] direction to form a ribbon structure.
Supporting information
CCDC reference: 296625
Key indicators
- Single-crystal X-ray study
- T = 93 K
- Mean
![[sigma]](https://journals.iucr.org/logos/entities/sigma_rmgif.gif) (C-C) = 0.004 Å
(C-C) = 0.004 Å
- R factor = 0.057
- wR factor = 0.172
- Data-to-parameter ratio = 10.5
checkCIF/PLATON results
No syntax errors found
 Alert level A
PLAT029_ALERT_3_A _diffrn_measured_fraction_theta_full Low ....... 0.93
Alert level A
PLAT029_ALERT_3_A _diffrn_measured_fraction_theta_full Low ....... 0.93
| Author Response: A two-dimensional detector (IP) was used in combination with Cu as
radiation.
|
 Alert level C
REFLT03_ALERT_3_C Reflection count < 95% complete
From the CIF: _diffrn_reflns_theta_max 68.22
From the CIF: _diffrn_reflns_theta_full 68.22
From the CIF: _reflns_number_total 1254
TEST2: Reflns within _diffrn_reflns_theta_max
Count of symmetry unique reflns 1347
Completeness (_total/calc) 93.10%
PLAT022_ALERT_3_C Ratio Unique / Expected Reflections too Low .... 0.93
Alert level C
REFLT03_ALERT_3_C Reflection count < 95% complete
From the CIF: _diffrn_reflns_theta_max 68.22
From the CIF: _diffrn_reflns_theta_full 68.22
From the CIF: _reflns_number_total 1254
TEST2: Reflns within _diffrn_reflns_theta_max
Count of symmetry unique reflns 1347
Completeness (_total/calc) 93.10%
PLAT022_ALERT_3_C Ratio Unique / Expected Reflections too Low .... 0.93
1 ALERT level A = In general: serious problem
0 ALERT level B = Potentially serious problem
2 ALERT level C = Check and explain
0 ALERT level G = General alerts; check
0 ALERT type 1 CIF construction/syntax error, inconsistent or missing data
0 ALERT type 2 Indicator that the structure model may be wrong or deficient
3 ALERT type 3 Indicator that the structure quality may be low
0 ALERT type 4 Improvement, methodology, query or suggestion
Compound (I) was purchased form Dainippon Ink & Chemicals Inc. and purified twice by sublimation using a two-zone furnace (Mizuguchi, 1981). Single crystals were grown from the vapour phase in a closed system at about 743 K. After 24 h, a number of red platelet [Needle below?] single crystals were obtained.
The H atom of the NH group was found in a difference density map and fixed during the refinement [Uiso(H) = 0.021 Å2]. All other H atoms were positioned geometrically and constrained to ride on their parent atoms, with C—H = 0.95 Å and Uiso(H) = 1.2Ueq(C).
Data collection: PROCESS-AUTO (Rigaku, 1998); cell refinement: PROCESS-AUTO; data reduction: CrystalStructure (Rigaku/MSC & Rigaku, 2005); program(s) used to solve structure: SHELXS86 (Sheldrick, 1986); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics: ORTEPIII (Burnett & Johnson, 1996); software used to prepare material for publication: CrystalStructure.
3,10-Dichloro-5,12-dihydroquino[2,3-
b]acridine-7,14-dione
top
Crystal data top
| C20H10Cl2N2O2 | Z = 1 |
| Mr = 381.20 | F(000) = 194.00 |
| Triclinic, P1 | Dx = 1.732 Mg m−3 |
| Hall symbol: -P 1 | Cu Kα radiation, λ = 1.5418 Å |
| a = 3.7635 (13) Å | Cell parameters from 2796 reflections |
| b = 5.853 (2) Å | θ = 10.7–136.4° |
| c = 16.746 (6) Å | µ = 4.17 mm−1 |
| α = 85.20 (2)° | T = 93 K |
| β = 83.79 (2)° | Needle, red |
| γ = 89.32 (2)° | 0.10 × 0.05 × 0.02 mm |
| V = 365.4 (2) Å3 | |
Data collection top
Rigaku R-AXIS RAPID imaging-plate
diffractometer | 1103 reflections with F2 > 2σ(F2) |
| Detector resolution: 10.00 pixels mm-1 | Rint = 0.046 |
| 72 frames, Δω = 10° scans | θmax = 68.2° |
Absorption correction: multi-scan
(ABSCOR; Higashi, 1995) | h = −4→4 |
| Tmin = 0.681, Tmax = 0.921 | k = −7→6 |
| 3173 measured reflections | l = −20→20 |
| 1254 independent reflections | |
Refinement top
| Refinement on F2 | H-atom parameters constrained |
| R[F2 > 2σ(F2)] = 0.057 | w = 1/[σ2(Fo2) + (0.1114P)2 + 0.1855P]
where P = (Fo2 + 2Fc2)/3 |
| wR(F2) = 0.172 | (Δ/σ)max < 0.001 |
| S = 1.13 | Δρmax = 0.60 e Å−3 |
| 1254 reflections | Δρmin = −0.38 e Å−3 |
| 119 parameters | |
Crystal data top
| C20H10Cl2N2O2 | γ = 89.32 (2)° |
| Mr = 381.20 | V = 365.4 (2) Å3 |
| Triclinic, P1 | Z = 1 |
| a = 3.7635 (13) Å | Cu Kα radiation |
| b = 5.853 (2) Å | µ = 4.17 mm−1 |
| c = 16.746 (6) Å | T = 93 K |
| α = 85.20 (2)° | 0.10 × 0.05 × 0.02 mm |
| β = 83.79 (2)° | |
Data collection top
Rigaku R-AXIS RAPID imaging-plate
diffractometer | 1254 independent reflections |
Absorption correction: multi-scan
(ABSCOR; Higashi, 1995) | 1103 reflections with F2 > 2σ(F2) |
| Tmin = 0.681, Tmax = 0.921 | Rint = 0.046 |
| 3173 measured reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.057 | 119 parameters |
| wR(F2) = 0.172 | H-atom parameters constrained |
| S = 1.13 | Δρmax = 0.60 e Å−3 |
| 1254 reflections | Δρmin = −0.38 e Å−3 |
Special details top
Geometry. All standard uncertainties (s.u.s) are estimated using the full covariance matrix. The cell s.u.s are taken into account individually in the estimation of s.u.s in distances and angles; correlations between s.u.s in cell parameters are only used when they are defined by crystal symmetry. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| Cl1 | 0.3744 (2) | 0.20197 (14) | 0.06111 (4) | 0.0274 (4) | |
| O1 | −0.3270 (6) | 0.8901 (4) | 0.33978 (12) | 0.0173 (6) | |
| N1 | 0.2179 (7) | 0.2827 (5) | 0.36148 (15) | 0.0160 (6) | |
| C1 | −0.0590 (8) | 0.6790 (6) | 0.19657 (18) | 0.0164 (7) | |
| H1 | −0.1733 | 0.8234 | 0.1886 | 0.020* | |
| C2 | 0.0589 (8) | 0.5644 (6) | 0.13156 (19) | 0.0200 (7) | |
| H2 | 0.0297 | 0.6276 | 0.0785 | 0.024* | |
| C3 | 0.2245 (8) | 0.3509 (6) | 0.14436 (18) | 0.0189 (7) | |
| C4 | 0.2763 (8) | 0.2534 (6) | 0.21913 (18) | 0.0175 (7) | |
| H4 | 0.3878 | 0.1076 | 0.2259 | 0.021* | |
| C5 | 0.1585 (8) | 0.3764 (5) | 0.28627 (18) | 0.0164 (7) | |
| C6 | −0.0155 (8) | 0.5880 (5) | 0.27631 (18) | 0.0153 (7) | |
| C7 | −0.1521 (8) | 0.7105 (6) | 0.34532 (18) | 0.0167 (7) | |
| C8 | −0.0715 (8) | 0.6025 (5) | 0.42458 (18) | 0.0150 (7) | |
| C9 | 0.1095 (8) | 0.3899 (5) | 0.43037 (18) | 0.0147 (7) | |
| C10 | −0.1758 (8) | 0.7107 (6) | 0.49436 (19) | 0.0166 (7) | |
| H10 | −0.2938 | 0.8549 | 0.4906 | 0.020* | |
| H1N | 0.3272 | 0.1676 | 0.3677 | 0.021* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| Cl1 | 0.0322 (6) | 0.0306 (6) | 0.0200 (5) | 0.0049 (4) | −0.0016 (3) | −0.0067 (3) |
| O1 | 0.0191 (11) | 0.0152 (13) | 0.0175 (11) | 0.0045 (10) | −0.0031 (9) | −0.0003 (8) |
| N1 | 0.0177 (13) | 0.0149 (14) | 0.0158 (14) | 0.0034 (11) | −0.0027 (10) | −0.0023 (10) |
| C1 | 0.0142 (15) | 0.0175 (17) | 0.0171 (15) | −0.0004 (13) | −0.0028 (12) | 0.0017 (12) |
| C2 | 0.0183 (16) | 0.0256 (19) | 0.0158 (16) | −0.0039 (14) | −0.0035 (12) | 0.0025 (12) |
| C3 | 0.0147 (15) | 0.0249 (19) | 0.0178 (16) | −0.0014 (14) | −0.0014 (12) | −0.0056 (13) |
| C4 | 0.0151 (15) | 0.0183 (17) | 0.0192 (16) | 0.0016 (14) | −0.0009 (12) | −0.0037 (12) |
| C5 | 0.0133 (15) | 0.0187 (18) | 0.0173 (15) | −0.0028 (13) | −0.0020 (12) | −0.0013 (12) |
| C6 | 0.0126 (15) | 0.0149 (17) | 0.0186 (17) | 0.0000 (13) | −0.0024 (12) | −0.0024 (12) |
| C7 | 0.0131 (15) | 0.0188 (18) | 0.0185 (16) | −0.0053 (13) | −0.0017 (12) | −0.0024 (12) |
| C8 | 0.0111 (15) | 0.0142 (17) | 0.0202 (16) | −0.0021 (13) | −0.0024 (12) | −0.0021 (12) |
| C9 | 0.0111 (14) | 0.0134 (17) | 0.0200 (16) | 0.0000 (13) | −0.0018 (12) | −0.0031 (12) |
| C10 | 0.0147 (15) | 0.0144 (16) | 0.0214 (17) | −0.0015 (13) | −0.0036 (12) | −0.0028 (12) |
Geometric parameters (Å, º) top
| Cl1—C3 | 1.744 (3) | C3—C4 | 1.365 (4) |
| O1—C7 | 1.236 (4) | C4—C5 | 1.414 (4) |
| N1—C5 | 1.371 (4) | C4—H4 | 0.9500 |
| N1—C9 | 1.380 (4) | C5—C6 | 1.400 (5) |
| N1—H1N | 0.7904 | C6—C7 | 1.454 (4) |
| C1—C2 | 1.357 (5) | C7—C8 | 1.482 (4) |
| C1—C6 | 1.421 (4) | C8—C10 | 1.392 (4) |
| C1—H1 | 0.9500 | C8—C9 | 1.413 (5) |
| C2—C3 | 1.400 (5) | C9—C10i | 1.392 (5) |
| C2—H2 | 0.9500 | C10—H10 | 0.9500 |
| | | |
| C5—N1—C9 | 122.2 (3) | C6—C5—C4 | 120.9 (3) |
| C5—N1—H1N | 121.8 | C5—C6—C1 | 118.0 (3) |
| C9—N1—H1N | 115.9 | C5—C6—C7 | 121.2 (3) |
| C2—C1—C6 | 121.6 (3) | C1—C6—C7 | 120.7 (3) |
| C2—C1—H1 | 119.2 | O1—C7—C6 | 123.5 (3) |
| C6—C1—H1 | 119.2 | O1—C7—C8 | 121.4 (3) |
| C1—C2—C3 | 118.5 (3) | C6—C7—C8 | 115.1 (3) |
| C1—C2—H2 | 120.7 | C10—C8—C9 | 119.4 (3) |
| C3—C2—H2 | 120.7 | C10—C8—C7 | 119.9 (3) |
| C4—C3—C2 | 123.1 (3) | C9—C8—C7 | 120.7 (3) |
| C4—C3—Cl1 | 118.1 (3) | N1—C9—C10i | 120.4 (3) |
| C2—C3—Cl1 | 118.8 (2) | N1—C9—C8 | 119.8 (3) |
| C3—C4—C5 | 117.8 (3) | C10i—C9—C8 | 119.7 (3) |
| C3—C4—H4 | 121.1 | C9i—C10—C8 | 120.9 (3) |
| C5—C4—H4 | 121.1 | C9i—C10—H10 | 119.6 |
| N1—C5—C6 | 120.8 (3) | C8—C10—H10 | 119.6 |
| N1—C5—C4 | 118.3 (3) | | |
| Symmetry code: (i) −x, −y+1, −z+1. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1N···O1ii | 0.79 | 2.12 | 2.873 (4) | 159 |
| Symmetry code: (ii) x+1, y−1, z. |
Experimental details
| Crystal data |
| Chemical formula | C20H10Cl2N2O2 |
| Mr | 381.20 |
| Crystal system, space group | Triclinic, P1 |
| Temperature (K) | 93 |
| a, b, c (Å) | 3.7635 (13), 5.853 (2), 16.746 (6) |
| α, β, γ (°) | 85.20 (2), 83.79 (2), 89.32 (2) |
| V (Å3) | 365.4 (2) |
| Z | 1 |
| Radiation type | Cu Kα |
| µ (mm−1) | 4.17 |
| Crystal size (mm) | 0.10 × 0.05 × 0.02 |
| |
| Data collection |
| Diffractometer | Rigaku R-AXIS RAPID imaging-plate
diffractometer |
| Absorption correction | Multi-scan
(ABSCOR; Higashi, 1995) |
| Tmin, Tmax | 0.681, 0.921 |
No. of measured, independent and
observed [F2 > 2σ(F2)] reflections | 3173, 1254, 1103 |
| Rint | 0.046 |
| (sin θ/λ)max (Å−1) | 0.602 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.057, 0.172, 1.13 |
| No. of reflections | 1254 |
| No. of parameters | 119 |
| No. of restraints | ? |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.60, −0.38 |
Selected bond lengths (Å) top| Cl1—C3 | 1.744 (3) | C4—C5 | 1.414 (4) |
| O1—C7 | 1.236 (4) | C5—C6 | 1.400 (5) |
| N1—C5 | 1.371 (4) | C6—C7 | 1.454 (4) |
| N1—C9 | 1.380 (4) | C7—C8 | 1.482 (4) |
| C1—C2 | 1.357 (5) | C8—C10 | 1.392 (4) |
| C1—C6 | 1.421 (4) | C8—C9 | 1.413 (5) |
| C2—C3 | 1.400 (5) | C9—C10i | 1.392 (5) |
| C3—C4 | 1.365 (4) | | |
| Symmetry code: (i) −x, −y+1, −z+1. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1N···O1ii | 0.79 | 2.12 | 2.873 (4) | 159 |
| Symmetry code: (ii) x+1, y−1, z. |

 Subscribe to Acta Crystallographica Section E: Crystallographic Communications
Subscribe to Acta Crystallographica Section E: Crystallographic Communications
The full text of this article is available to subscribers to the journal.
If you have already registered and are using a computer listed in your registration details, please email
[email protected] for assistance.


 journal menu
journal menu organic compounds
organic compounds












![[Figure 1]](https://journals.iucr.org/e/issues/2006/01/00/ob6599/ob6599fig1thm.gif)
![[Figure 2]](https://journals.iucr.org/e/issues/2006/01/00/ob6599/ob6599fig2thm.gif)




The title compound, (I), abbreviated to 3,10-DClQA, is one of the quinacridone derivatives on the market which are known as red pigments (Pigment Red 209; Herbst & Hunger, 1997) and typically characterized by N—H···O intermolecular hydrogen bonds. 3,10-DClQA exhibits a yellowish–red shade in the solid state, while the colour is bluish–red in 2,9-dichloroquinacridone (2,9-DClQA; Senju et al., 2005), although the solution spectra of both compounds are practically the same. This suggests that intermolecular interactions in the solid state are responsible for the difference in colour. In this connection, the structure analysis of (I) has been undertaken.
Fig. 1 shows a plot of the molecule of (I). The molecule has inversion symmetry. The quinacridine skeleton is entirely planar, as indicated by a small deviation [For which atom?] of about 0.02 Å from the least-squares plane of the rings defined by atoms C1–C10 and N1. However, the carbonyl O atom deviates by 0.140 (3) Å from the least-squares plane of the ring system towards the NH group of the neighbouring molecule, enabling the formation of an N—H···O intermolecular hydrogen bond (Table 2). This tendency was also found in 2,9-DClQA (Senju et al., 2005).
As shown in Fig. 2(a), there are chains of N—H···O intermolecular hydrogen bonds along the [110] direction. One molecule is bonded to two neighbouring molecules through four hydrogen bonds. There is a small step of about 0.55 Å between the two molecular planes of the hydrogen-bonded molecules, as shown in Fig. 2(b). In commercial hydrogen-bonded pigments, there are normally no steps between molecules (Mizuguchi et al., 1992, 1993) and this is a good criterion for strong hydrogen bonds. The existence of the step in (I) indicates a somewhat weaker hydrogen bond. This kind of step has also been found in the following pigments: 2,9-dimethylquinacridone (Mizuguchi et al., 2002), modifications I and II of dithioketopyrrolopyrrole (Mizuguchi et al., 1990), thiazine-indigo (Senju & Mizuguchi, 2003) and 2,9-DClQA (Senju et al., 2005). In the present investigation, no significant difference in structure has been observed between 3,10-DClQA [(I)] and 2,9-DClQA, contrary to our expectation. Further investigation is now in progress in order to elucidate the origin of the difference in colour in the solid state.