2,2′-Bipyridine (2BPY) and hexahydroxybenzene (HHB) crystallize in a 2:1 ratio as a neutral molecular adduct, C
6H
6O
6·2C
10H
8N
2, in space group
P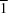
with
Z = 1 and with the HHB molecule lying on an inversion centre. HHB, of which this is the first single-crystal X-ray structure determination, forms O—H

O hydrogen-bonded chains parallel to the
a axis, with O
O distances of 2.761 (1) and 2.782 (1) Å. O—H
N hydrogen bonds to the 2BPY molecules crosslink these chains, with O
N distances of 2.707 (1) and 2.735 (1) Å.
Supporting information
CCDC reference: 174836
A solution of 2,2'-bipyridine in methanol (0.5 ml, 0.2M) was poured
slowly into a narrow test tube containing an aqueous solution of
tetrahydroxybenzoquinone (approximately 0.5 ml, 0.2 M) to produce an
interface. Pale-yellow plate crystals of (I) suitable for X-ray analysis were
produced by slow mixing of the solutions. IR spectra were recorded on the
starting materials and product using KBr discs on a Perkin-Elmer 580B IR
spectrometer.
All H atoms were located in difference Fourier maps and refined with isotropic
displacement parameters; the C—H and O—H distances all refined to within
standard ranges and there were no anomalous values of Uiso. The
highest peak in the residual electron density is 0.40 e Å-3 and the ten
highest peaks all lie in the centres of aromatic bonds. There is no indication
of disorder in the O—H hydrogen bonding.
Data collection: SMART (Bruker, 1998); cell refinement: SMART; data reduction: SAINT (Bruker, 1998); program(s) used to solve structure: SHELXS97 (Sheldrick, 1990); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics: SHELXTL (Sheldrick, 1999); software used to prepare material for publication: SHELXTL.
Hexahydroxybenzene-2,2'-bipyridine (1/2)
top
Crystal data top
| C6H6O6·2C10H8N2 | Z = 1 |
| Mr = 486.48 | F(000) = 254 |
| Triclinic, P1 | Dx = 1.464 Mg m−3 |
| a = 7.2913 (5) Å | Mo Kα radiation, λ = 0.71073 Å |
| b = 7.9434 (5) Å | Cell parameters from 991 reflections |
| c = 9.9197 (7) Å | θ = 10.3–30.8° |
| α = 76.584 (4)° | µ = 0.11 mm−1 |
| β = 80.959 (4)° | T = 100 K |
| γ = 86.321 (4)° | Plate, pale yellow |
| V = 551.68 (6) Å3 | 0.36 × 0.32 × 0.12 mm |
Data collection top
Bruker SMART CCD area-detector
diffractometer | 2536 reflections with I > 2σ(I) |
| Radiation source: fine-focus sealed tube | Rint = 0.034 |
| Graphite monochromator | θmax = 29.0°, θmin = 2.1° |
| ω scans | h = −9→9 |
| 6594 measured reflections | k = −10→10 |
| 2850 independent reflections | l = −13→13 |
Refinement top
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.040 | Hydrogen site location: difference Fourier map |
| wR(F2) = 0.112 | All H-atom parameters refined |
| S = 1.06 | w = 1/[σ2(Fo2) + (0.0568P)2 + 0.196P]
where P = (Fo2 + 2Fc2)/3 |
| 2850 reflections | (Δ/σ)max < 0.001 |
| 207 parameters | Δρmax = 0.40 e Å−3 |
| 0 restraints | Δρmin = −0.20 e Å−3 |
Crystal data top
| C6H6O6·2C10H8N2 | γ = 86.321 (4)° |
| Mr = 486.48 | V = 551.68 (6) Å3 |
| Triclinic, P1 | Z = 1 |
| a = 7.2913 (5) Å | Mo Kα radiation |
| b = 7.9434 (5) Å | µ = 0.11 mm−1 |
| c = 9.9197 (7) Å | T = 100 K |
| α = 76.584 (4)° | 0.36 × 0.32 × 0.12 mm |
| β = 80.959 (4)° | |
Data collection top
Bruker SMART CCD area-detector
diffractometer | 2536 reflections with I > 2σ(I) |
| 6594 measured reflections | Rint = 0.034 |
| 2850 independent reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.040 | 0 restraints |
| wR(F2) = 0.112 | All H-atom parameters refined |
| S = 1.06 | Δρmax = 0.40 e Å−3 |
| 2850 reflections | Δρmin = −0.20 e Å−3 |
| 207 parameters | |
Special details top
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes)
are estimated using the full covariance matrix. The cell e.s.d.'s are taken
into account individually in the estimation of e.s.d.'s in distances, angles
and torsion angles; correlations between e.s.d.'s in cell parameters are only
used when they are defined by crystal symmetry. An approximate (isotropic)
treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s.
planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor
wR and goodness of fit S are based on F2, conventional
R-factors R are based on F, with F set to zero for
negative F2. The threshold expression of F2 >
σ(F2) is used only for calculating R-factors(gt) etc.
and is not relevant to the choice of reflections for refinement.
R-factors based on F2 are statistically about twice as large
as those based on F, and R- factors based on ALL data will be
even larger. As we are interested in the behaviour of the H atoms with respect to any
hydrogen-bonded networks, it would be artificial to add any constraints to
this stable and converged refinement. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| C1 | 0.16996 (15) | 0.52804 (13) | −0.08938 (11) | 0.0142 (2) | |
| O1 | 0.33398 (11) | 0.55394 (11) | −0.18105 (8) | 0.01782 (19) | |
| H1 | 0.427 (3) | 0.537 (3) | −0.136 (2) | 0.045 (5)* | |
| C2 | 0.16557 (15) | 0.44537 (13) | 0.05206 (11) | 0.0139 (2) | |
| O2 | 0.33287 (11) | 0.39335 (10) | 0.09993 (8) | 0.01630 (18) | |
| H2 | 0.315 (3) | 0.295 (3) | 0.163 (2) | 0.048 (5)* | |
| C3 | −0.00342 (15) | 0.41799 (13) | 0.14194 (10) | 0.0143 (2) | |
| O3 | 0.00197 (12) | 0.34774 (10) | 0.28181 (8) | 0.01730 (19) | |
| H3 | −0.077 (3) | 0.261 (3) | 0.315 (2) | 0.043 (5)* | |
| C10 | 0.26616 (15) | −0.04066 (14) | 0.37534 (11) | 0.0152 (2) | |
| N1 | 0.30727 (13) | 0.06018 (12) | 0.24539 (10) | 0.0169 (2) | |
| C12 | 0.31299 (17) | −0.01257 (15) | 0.13510 (12) | 0.0202 (2) | |
| H12 | 0.342 (2) | 0.067 (2) | 0.0444 (17) | 0.025 (4)* | |
| C13 | 0.27951 (18) | −0.18639 (15) | 0.14682 (13) | 0.0223 (2) | |
| H13 | 0.287 (2) | −0.231 (2) | 0.0611 (17) | 0.027 (4)* | |
| C14 | 0.23513 (18) | −0.28999 (15) | 0.28074 (12) | 0.0209 (2) | |
| H14 | 0.204 (2) | −0.413 (2) | 0.2928 (18) | 0.033 (4)* | |
| C15 | 0.22906 (17) | −0.21697 (14) | 0.39622 (12) | 0.0184 (2) | |
| H15 | 0.198 (2) | −0.289 (2) | 0.4884 (18) | 0.030 (4)* | |
| C20 | 0.26165 (15) | 0.04352 (13) | 0.49592 (11) | 0.0153 (2) | |
| N2 | 0.19573 (14) | −0.05152 (12) | 0.62402 (10) | 0.0177 (2) | |
| C22 | 0.18863 (18) | 0.01969 (15) | 0.73523 (12) | 0.0211 (2) | |
| H22 | 0.141 (2) | −0.054 (2) | 0.8248 (18) | 0.028 (4)* | |
| C23 | 0.24377 (18) | 0.18730 (16) | 0.72545 (12) | 0.0213 (2) | |
| H23 | 0.232 (2) | 0.231 (2) | 0.8112 (18) | 0.031 (4)* | |
| C24 | 0.31240 (17) | 0.28460 (15) | 0.59350 (13) | 0.0205 (2) | |
| H24 | 0.356 (2) | 0.402 (2) | 0.5849 (17) | 0.030 (4)* | |
| C25 | 0.32362 (17) | 0.21210 (14) | 0.47730 (12) | 0.0190 (2) | |
| H25 | 0.376 (2) | 0.275 (2) | 0.3857 (17) | 0.027 (4)* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| C1 | 0.0153 (5) | 0.0133 (4) | 0.0139 (4) | −0.0024 (4) | 0.0003 (4) | −0.0039 (4) |
| O1 | 0.0139 (4) | 0.0231 (4) | 0.0149 (4) | −0.0027 (3) | 0.0000 (3) | −0.0020 (3) |
| C2 | 0.0149 (5) | 0.0124 (4) | 0.0150 (5) | −0.0018 (4) | −0.0029 (4) | −0.0032 (4) |
| O2 | 0.0149 (4) | 0.0157 (4) | 0.0168 (4) | −0.0021 (3) | −0.0035 (3) | 0.0006 (3) |
| C3 | 0.0175 (5) | 0.0130 (4) | 0.0122 (4) | −0.0021 (4) | −0.0016 (4) | −0.0022 (3) |
| O3 | 0.0195 (4) | 0.0188 (4) | 0.0125 (4) | −0.0046 (3) | −0.0024 (3) | −0.0001 (3) |
| C10 | 0.0148 (5) | 0.0150 (5) | 0.0159 (5) | −0.0005 (4) | −0.0019 (4) | −0.0037 (4) |
| N1 | 0.0168 (4) | 0.0161 (4) | 0.0168 (4) | −0.0004 (3) | −0.0008 (3) | −0.0030 (3) |
| C12 | 0.0243 (6) | 0.0197 (5) | 0.0158 (5) | −0.0006 (4) | −0.0010 (4) | −0.0034 (4) |
| C13 | 0.0283 (6) | 0.0206 (5) | 0.0192 (5) | −0.0015 (4) | −0.0017 (4) | −0.0080 (4) |
| C14 | 0.0250 (6) | 0.0166 (5) | 0.0216 (5) | −0.0024 (4) | −0.0015 (4) | −0.0063 (4) |
| C15 | 0.0217 (5) | 0.0149 (5) | 0.0177 (5) | −0.0016 (4) | −0.0009 (4) | −0.0032 (4) |
| C20 | 0.0153 (5) | 0.0144 (5) | 0.0163 (5) | 0.0009 (4) | −0.0032 (4) | −0.0034 (4) |
| N2 | 0.0194 (5) | 0.0170 (4) | 0.0165 (4) | −0.0016 (4) | −0.0026 (4) | −0.0030 (3) |
| C22 | 0.0252 (6) | 0.0216 (5) | 0.0161 (5) | −0.0027 (4) | −0.0021 (4) | −0.0037 (4) |
| C23 | 0.0249 (6) | 0.0213 (5) | 0.0200 (5) | 0.0007 (4) | −0.0053 (4) | −0.0083 (4) |
| C24 | 0.0235 (6) | 0.0162 (5) | 0.0230 (5) | −0.0008 (4) | −0.0061 (4) | −0.0049 (4) |
| C25 | 0.0221 (6) | 0.0156 (5) | 0.0190 (5) | −0.0023 (4) | −0.0039 (4) | −0.0026 (4) |
Geometric parameters (Å, º) top
| C1—O1 | 1.3793 (13) | C13—C14 | 1.3923 (16) |
| C1—C3i | 1.3994 (15) | C13—H13 | 0.985 (16) |
| C1—C2 | 1.4007 (14) | C14—C15 | 1.3928 (16) |
| O1—H1 | 0.86 (2) | C14—H14 | 0.991 (17) |
| C2—O2 | 1.3822 (13) | C15—H15 | 0.961 (17) |
| C2—C3 | 1.3999 (15) | C20—N2 | 1.3509 (14) |
| O2—H2 | 0.88 (2) | C20—C25 | 1.4029 (15) |
| C3—O3 | 1.3758 (12) | N2—C22 | 1.3445 (15) |
| C3—C1i | 1.3994 (15) | C22—C23 | 1.3929 (17) |
| O3—H3 | 0.89 (2) | C22—H22 | 0.968 (17) |
| C10—N1 | 1.3503 (14) | C23—C24 | 1.3912 (17) |
| C10—C15 | 1.4049 (15) | C23—H23 | 0.978 (17) |
| C10—C20 | 1.4950 (15) | C24—C25 | 1.3926 (16) |
| N1—C12 | 1.3448 (15) | C24—H24 | 0.986 (17) |
| C12—C13 | 1.3926 (16) | C25—H25 | 0.961 (17) |
| C12—H12 | 0.975 (16) | | |
| | | |
| O1—C1—C2 | 122.08 (10) | C13—C14—C15 | 119.06 (11) |
| O1—C1—C3i | 118.22 (9) | C13—C14—H14 | 120.0 (10) |
| C3i—C1—C2 | 119.66 (10) | C15—C14—H14 | 120.9 (10) |
| C1—O1—H1 | 110.3 (14) | C14—C15—C10 | 119.50 (10) |
| O2—C2—C1 | 117.96 (9) | C14—C15—H15 | 118.7 (10) |
| O2—C2—C3 | 121.28 (9) | C10—C15—H15 | 121.8 (10) |
| C3—C2—C1 | 120.76 (10) | N2—C20—C25 | 121.66 (10) |
| C2—O2—H2 | 107.6 (14) | N2—C20—C10 | 116.55 (9) |
| O3—C3—C1i | 122.28 (9) | C25—C20—C10 | 121.79 (10) |
| O3—C3—C2 | 118.03 (10) | C22—N2—C20 | 118.43 (10) |
| C1i—C3—C2 | 119.58 (9) | N2—C22—C23 | 123.51 (11) |
| C3—O3—H3 | 111.2 (13) | N2—C22—H22 | 115.5 (10) |
| N1—C10—C15 | 121.41 (10) | C23—C22—H22 | 121.0 (10) |
| N1—C10—C20 | 117.11 (9) | C24—C23—C22 | 117.94 (11) |
| C15—C10—C20 | 121.48 (10) | C24—C23—H23 | 123.5 (10) |
| C12—N1—C10 | 118.29 (10) | C22—C23—H23 | 118.6 (10) |
| N1—C12—C13 | 123.86 (10) | C23—C24—C25 | 119.39 (11) |
| N1—C12—H12 | 114.1 (10) | C23—C24—H24 | 119.1 (10) |
| C13—C12—H12 | 122.0 (10) | C25—C24—H24 | 121.5 (10) |
| C14—C13—C12 | 117.87 (11) | C24—C25—C20 | 119.05 (10) |
| C14—C13—H13 | 122.9 (10) | C24—C25—H25 | 121.1 (10) |
| C12—C13—H13 | 119.2 (10) | C20—C25—H25 | 119.9 (10) |
| Symmetry code: (i) −x, −y+1, −z. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| O1—H1···O2ii | 0.86 (2) | 1.98 (2) | 2.761 (1) | 151 (2) |
| O1—H1···O2 | 0.86 (2) | 2.37 (2) | 2.782 (1) | 110 (2) |
| O2—H2···N1 | 0.88 (2) | 1.86 (2) | 2.707 (1) | 162 (2) |
| O3—H3···N2iii | 0.89 (2) | 1.86 (2) | 2.735 (1) | 168 (2) |
| C14—H14···O3iv | 0.99 (2) | 2.51 (2) | 3.432 (1) | 154 (14) |
| C15—H15···O3iii | 0.96 (2) | 2.47 (2) | 3.333 (1) | 149 (14) |
| Symmetry codes: (ii) −x+1, −y+1, −z; (iii) −x, −y, −z+1; (iv) x, y−1, z. |
Experimental details
| Crystal data |
| Chemical formula | C6H6O6·2C10H8N2 |
| Mr | 486.48 |
| Crystal system, space group | Triclinic, P1 |
| Temperature (K) | 100 |
| a, b, c (Å) | 7.2913 (5), 7.9434 (5), 9.9197 (7) |
| α, β, γ (°) | 76.584 (4), 80.959 (4), 86.321 (4) |
| V (Å3) | 551.68 (6) |
| Z | 1 |
| Radiation type | Mo Kα |
| µ (mm−1) | 0.11 |
| Crystal size (mm) | 0.36 × 0.32 × 0.12 |
| |
| Data collection |
| Diffractometer | Bruker SMART CCD area-detector
diffractometer |
| Absorption correction | – |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 6594, 2850, 2536 |
| Rint | 0.034 |
| (sin θ/λ)max (Å−1) | 0.682 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.040, 0.112, 1.06 |
| No. of reflections | 2850 |
| No. of parameters | 207 |
| H-atom treatment | All H-atom parameters refined |
| Δρmax, Δρmin (e Å−3) | 0.40, −0.20 |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| O1—H1···O2i | 0.86 (2) | 1.98 (2) | 2.761 (1) | 151 (2) |
| O1—H1···O2 | 0.86 (2) | 2.37 (2) | 2.782 (1) | 110 (2) |
| O2—H2···N1 | 0.88 (2) | 1.86 (2) | 2.707 (1) | 162 (2) |
| O3—H3···N2ii | 0.89 (2) | 1.86 (2) | 2.735 (1) | 168 (2) |
| C14—H14···O3iii | 0.99 (2) | 2.51 (2) | 3.432 (1) | 154 (14) |
| C15—H15···O3ii | 0.96 (2) | 2.47 (2) | 3.333 (1) | 149 (14) |
| Symmetry codes: (i) −x+1, −y+1, −z; (ii) −x, −y, −z+1; (iii) x, y−1, z. |


 journal menu
journal menu organic compounds
organic compounds











![[Figure 1]](https://journals.iucr.org/c/issues/2001/10/00/gg1065/gg1065fig1thm.gif)
![[Figure 2]](https://journals.iucr.org/c/issues/2001/10/00/gg1065/gg1065fig2thm.gif)




In order to investigate a variety of molecular interactions in the solid state, in particular N—H···O and O—H···N hydrogen bonds, we attempted to produce co-crystals of 2,2'-bipyridine (2BPY) and tetrahydroxybenzoquinone (THBQ), but during the crystallization process the THBQ was reduced to hexahydroxybenzene (HHB). The resulting co-crystal, (I), comprised a neutral molecular adduct of 2BPY and HHB in a 2:1 ratio in space group P1 with Z = 1, the HHB residing on an inversion centre. \sch
Reduction of quinone to hydroquinone is a reaction commonly found in organic text books (March, 1985; Vollhardt & Schore, 1994). The addition of hydroxyl groups to the ring weakens the carbonyl bond in quinone derivatives, as can be seen in the X-ray diffraction-determined bond lengths in quinone (C═O 1.222 Å; Allen et al., 1992), 2,5-dihydroxybenzoquinone, (C═O 1.232 Å; Cowan et al., 2001a) and THBQ (C═O 1.235 Å; Cowan et al., 2001b). This weakening of the carbonyl bond facilitates reduction by stabilizing the reaction intermediates (Vollhardt & Schore, 1994). Indeed, HHB is synthesized from THBQ (Fatiadi & Sager, 1975) but in acidic conditions; we are unsure how this reaction has taken place in the essentially neutral conditions of the present crystallization.
IR absorption spectra were recorded from the crystal of (I), from THBQ, and from a 2:1 mixture of 2BPY and THBQ. The spectra of THBQ and of the THBQ and 2BPY mixture show a significant carbonyl peak at 1600 cm-1, while the spectrum from (I) shows no significant peak in this region. The freshly prepared KBr disc of (I) was colourless, but it turned pale pink a few hours after preparation, which is evidence of moisture- and air-induced oxidation of the HHB to TBHQ, as observed by Fatiadi & Sager (1975) in pure HHB crystals. THBQ is highly coloured and a trace amount can cause intense colouration. After recollection of the spectrum on the reprepared discoloured disc of (I), no significant difference was observed.
The HHB molecular refinement behaves normally and there is no evidence in the s.u.s of the bond lengths, or in unusual anisotropic displacement parameters, of rotational disorder that could diguize THBQ as HHB (Fig. 1). The molecular geometry of the HHB is as expected, although there are no examples in the Cambridge Structural Database (CSD; Allen & Kennard, 1993) for comparison. All the C—C bond distances are identical to within three s.u.s [1.399 (2), 1.401 (1) and 1.399 (2) Å] and there are only slight differences in the C—O bond distances [1.379 (1), 1.382 (1) and 1.376 (2) Å]. The average C—C—O angles of the hydroxy groups to the benzene ring are 118.1° trans to the H atom and 121.9° cis to the H atom. This asymmetry is common in phenol molecules in the CSD and is due to the asymmetry of the hydroxyl group. The 2BPY moiety can be neutral, or singly or doubly protonated. In this case it is unprotonated and in an almost planar trans conformation; the N1—C10—C20—N2 torsion angle is 171.15 (10)°, with an angle of 8.73 (6)° between the two pyridyl rings.
The HHB molecules, which occupy centrosymmetric unit-cell corner sites, form columns of parallel rings in the b direction and hydrogen-bonded chains in the a direction [O1···O2i 2.761 (1) Å; symmetry code: (i) 1 - x, 1 - y, -z]. The 2BPY molecules, which stack in pairs with a mean separation of 3.46 Å, lie approximately parallel to the bc plane and these stacks form zigzag columns in the a direction, with a separation between 2BPY `dimers' of 3.50 Å. A dimer of 2BPY molecules is hydrogen bonded to the same HHB molecule, with O···N distances of O2···N1 2.707 (1) Å and O3···N2ii 2.735 (1) Å linking the HHB columns to the 2BPY dimers [symmetry code: (ii) -x, -y, 1 - z; Fig. 2]. The 2BPY and HHB molecules in (I) are also linked by two weak C···O hydrogen bonds, with C15···O3ii 3.333 (1) Å and C14···O3iii 3.432 (1) Å [symmetry code: (iii) x, y - 1, z]; details are given in Table 1.