In the title compound, C
6H
18N
22+·2C
2H
2ClO
2−, the cation lies across an inversion centre in the
P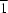
space group. The ions are linked by two two-centre N—H

O hydrogen bonds and by one three-centre N—H
(O)
2 hydrogen bond to form a three-dimensional framework structure. The significance of this study lies in the analysis of the complex hydrogen-bonded structure and in the comparison of this structure with those of other simple hexamethylenediammonium salts.
Supporting information
CCDC reference: 703734
A solution of hexamethylenediamine (4.0 g, 0.03 mmol) in a mixture of water (2 ml) and ethanol (4 ml) was cooled to 273 K, chloroacetic acid (6.5 g, 0.06 mmol) was added in small portions, and the mixture was stirred at room
temperature for 4 h. The resulting solution was added to acetone (20 ml),
producing a colourless crystalline precipitate of (I). This product was
collected by filtration and dried, giving crystals suitable for single-crystal
X-ray diffraction (yield 84%; m.p. > 623 K).
All H atoms were located in difference maps and then treated as riding atoms in
geometrically idealized positions, with C—H 0.99 Å and N—H 0.88 Å, and
with Uiso(H) = 1.2Ueq(carrier).
Data collection: COLLECT (Nonius, 1999); cell refinement: DIRAX/LSQ (Duisenberg et al., 2000); data reduction: EVALCCD (Duisenberg et al., 2003); program(s) used to solve structure: SIR2004 (Burla et al., 2005); program(s) used to refine structure: OSCAIL (McArdle, 2003) and SHELXL97 (Sheldrick, 2008); molecular graphics: PLATON (Spek, 2003); software used to prepare material for publication: SHELXL97 (Sheldrick, 2008) and PRPKAPPA (Ferguson, 1999).
Hexamethylenediammonium bis(chloroacetate)
top
Crystal data top
| C6H18N22+·2C2H2ClO2− | Z = 1 |
| Mr = 305.20 | F(000) = 162 |
| Triclinic, P1 | Dx = 1.359 Mg m−3 |
| Hall symbol: -P 1 | Mo Kα radiation, λ = 0.71073 Å |
| a = 6.5779 (18) Å | Cell parameters from 1718 reflections |
| b = 7.3593 (10) Å | θ = 3.0–27.5° |
| c = 8.425 (2) Å | µ = 0.44 mm−1 |
| α = 97.277 (12)° | T = 120 K |
| β = 95.631 (15)° | Lath, colourless |
| γ = 111.037 (12)° | 0.42 × 0.12 × 0.09 mm |
| V = 373.03 (15) Å3 | |
Data collection top
Bruker Nonius KappaCCD
diffractometer | 1718 independent reflections |
| Radiation source: Bruker Nonius FR591 rotating anode | 1273 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.044 |
| Detector resolution: 9.091 pixels mm-1 | θmax = 27.5°, θmin = 3.0° |
| ϕ and ω scans | h = −8→8 |
Absorption correction: multi-scan
(SADABS; Sheldrick, 2003) | k = −9→9 |
| Tmin = 0.836, Tmax = 0.961 | l = −10→10 |
| 10725 measured reflections | |
Refinement top
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.039 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.097 | H-atom parameters constrained |
| S = 1.09 | w = 1/[σ2(Fo2) + (0.0385P)2 + 0.2039P]
where P = (Fo2 + 2Fc2)/3 |
| 1718 reflections | (Δ/σ)max < 0.001 |
| 82 parameters | Δρmax = 0.28 e Å−3 |
| 0 restraints | Δρmin = −0.32 e Å−3 |
Crystal data top
| C6H18N22+·2C2H2ClO2− | γ = 111.037 (12)° |
| Mr = 305.20 | V = 373.03 (15) Å3 |
| Triclinic, P1 | Z = 1 |
| a = 6.5779 (18) Å | Mo Kα radiation |
| b = 7.3593 (10) Å | µ = 0.44 mm−1 |
| c = 8.425 (2) Å | T = 120 K |
| α = 97.277 (12)° | 0.42 × 0.12 × 0.09 mm |
| β = 95.631 (15)° | |
Data collection top
Bruker Nonius KappaCCD
diffractometer | 1718 independent reflections |
Absorption correction: multi-scan
(SADABS; Sheldrick, 2003) | 1273 reflections with I > 2σ(I) |
| Tmin = 0.836, Tmax = 0.961 | Rint = 0.044 |
| 10725 measured reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.039 | 0 restraints |
| wR(F2) = 0.097 | H-atom parameters constrained |
| S = 1.09 | Δρmax = 0.28 e Å−3 |
| 1718 reflections | Δρmin = −0.32 e Å−3 |
| 82 parameters | |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| Cl1 | 1.20850 (9) | 0.87484 (8) | 0.33120 (6) | 0.03979 (18) | |
| O1 | 0.8943 (2) | 0.72297 (18) | 0.02519 (15) | 0.0233 (3) | |
| O2 | 0.7035 (2) | 0.91290 (19) | 0.08073 (17) | 0.0278 (3) | |
| N1 | 0.7202 (2) | 0.3237 (2) | 0.05569 (18) | 0.0211 (3) | |
| C2 | 0.7302 (3) | 0.3420 (3) | 0.2332 (2) | 0.0235 (4) | |
| C3 | 0.5837 (3) | 0.4444 (3) | 0.2917 (2) | 0.0221 (4) | |
| C4 | 0.5738 (3) | 0.4491 (3) | 0.4704 (2) | 0.0231 (4) | |
| C5 | 1.0221 (3) | 0.9807 (3) | 0.2583 (2) | 0.0264 (4) | |
| C6 | 0.8592 (3) | 0.8592 (3) | 0.1085 (2) | 0.0199 (4) | |
| H1A | 0.7536 | 0.4411 | 0.0280 | 0.025* | |
| H1B | 0.5839 | 0.2509 | 0.0099 | 0.025* | |
| H1C | 0.8125 | 0.2704 | 0.0229 | 0.025* | |
| H2A | 0.8841 | 0.4182 | 0.2865 | 0.028* | |
| H2B | 0.6832 | 0.2089 | 0.2631 | 0.028* | |
| H3A | 0.4330 | 0.3752 | 0.2302 | 0.026* | |
| H3B | 0.6393 | 0.5815 | 0.2701 | 0.026* | |
| H4A | 0.5191 | 0.3120 | 0.4920 | 0.028* | |
| H4B | 0.7245 | 0.5188 | 0.5318 | 0.028* | |
| H5A | 0.9384 | 1.0015 | 0.3456 | 0.032* | |
| H5B | 1.1083 | 1.1121 | 0.2336 | 0.032* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| Cl1 | 0.0394 (3) | 0.0482 (3) | 0.0353 (3) | 0.0253 (3) | −0.0062 (2) | 0.0025 (2) |
| O1 | 0.0258 (7) | 0.0216 (7) | 0.0243 (7) | 0.0106 (5) | 0.0068 (5) | 0.0034 (5) |
| O2 | 0.0221 (7) | 0.0253 (7) | 0.0367 (8) | 0.0122 (6) | 0.0009 (6) | 0.0014 (6) |
| N1 | 0.0212 (8) | 0.0192 (8) | 0.0252 (8) | 0.0096 (6) | 0.0073 (6) | 0.0029 (6) |
| C2 | 0.0256 (9) | 0.0275 (10) | 0.0201 (9) | 0.0136 (8) | 0.0040 (7) | 0.0027 (7) |
| C3 | 0.0254 (9) | 0.0208 (9) | 0.0229 (9) | 0.0112 (8) | 0.0060 (7) | 0.0049 (7) |
| C4 | 0.0264 (9) | 0.0240 (9) | 0.0222 (9) | 0.0124 (8) | 0.0052 (7) | 0.0050 (7) |
| C5 | 0.0314 (10) | 0.0243 (10) | 0.0248 (9) | 0.0133 (8) | 0.0013 (8) | 0.0027 (8) |
| C6 | 0.0205 (9) | 0.0175 (8) | 0.0222 (9) | 0.0054 (7) | 0.0072 (7) | 0.0069 (7) |
Geometric parameters (Å, º) top
| N1—C2 | 1.477 (2) | C4—C4i | 1.509 (3) |
| N1—H1A | 0.88 | C4—H4A | 0.99 |
| N1—H1B | 0.88 | C4—H4B | 0.99 |
| N1—H1C | 0.88 | C5—C6 | 1.517 (3) |
| C2—C3 | 1.503 (2) | C5—H5A | 0.99 |
| C2—H2A | 0.99 | C5—H5B | 0.99 |
| C2—H2B | 0.99 | C5—Cl1 | 1.7742 (19) |
| C3—C4 | 1.510 (2) | C6—O1 | 1.253 (2) |
| C3—H3A | 0.99 | C6—O2 | 1.236 (2) |
| C3—H3B | 0.99 | | |
| | | |
| C2—N1—H1A | 109.7 | H3A—C3—H3B | 107.9 |
| C2—N1—H1C | 110.5 | C4i—C4—C3 | 112.50 (19) |
| H1A—N1—H1C | 109.6 | C4i—C4—H4A | 109.1 |
| C2—N1—H1B | 108.4 | C3—C4—H4A | 109.1 |
| H1A—N1—H1B | 107.7 | C4i—C4—H4B | 109.1 |
| H1B—N1—H1C | 110.9 | C3—C4—H4B | 109.1 |
| N1—C2—C3 | 110.88 (14) | H4A—C4—H4B | 107.8 |
| N1—C2—H2A | 109.5 | C6—C5—Cl1 | 115.00 (13) |
| C3—C2—H2A | 109.5 | C6—C5—H5A | 108.5 |
| N1—C2—H2B | 109.5 | Cl1—C5—H5A | 108.5 |
| C3—C2—H2B | 109.5 | C6—C5—H5B | 108.5 |
| H2A—C2—H2B | 108.1 | Cl1—C5—H5B | 108.5 |
| C2—C3—C4 | 112.26 (15) | H5A—C5—H5B | 107.5 |
| C2—C3—H3A | 109.2 | O2—C6—O1 | 126.87 (17) |
| C4—C3—H3A | 109.2 | O2—C6—C5 | 113.52 (15) |
| C2—C3—H3B | 109.2 | O1—C6—C5 | 119.60 (16) |
| C4—C3—H3B | 109.2 | | |
| | | |
| N1—C2—C3—C4 | 174.78 (15) | Cl1—C5—C6—O1 | −16.3 (2) |
| C2—C3—C4—C4i | −179.73 (19) | Cl1—C5—C6—O2 | 165.29 (13) |
| Symmetry code: (i) −x+1, −y+1, −z+1. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1A···O1 | 0.88 | 1.95 | 2.801 (2) | 163 |
| N1—H1B···O2ii | 0.88 | 1.86 | 2.739 (2) | 177 |
| N1—H1C···O1iii | 0.88 | 1.99 | 2.814 (2) | 154 |
| N1—H1C···O2iv | 0.88 | 2.59 | 3.020 (2) | 111 |
| Symmetry codes: (ii) −x+1, −y+1, −z; (iii) −x+2, −y+1, −z; (iv) x, y−1, z. |
Experimental details
| Crystal data |
| Chemical formula | C6H18N22+·2C2H2ClO2− |
| Mr | 305.20 |
| Crystal system, space group | Triclinic, P1 |
| Temperature (K) | 120 |
| a, b, c (Å) | 6.5779 (18), 7.3593 (10), 8.425 (2) |
| α, β, γ (°) | 97.277 (12), 95.631 (15), 111.037 (12) |
| V (Å3) | 373.03 (15) |
| Z | 1 |
| Radiation type | Mo Kα |
| µ (mm−1) | 0.44 |
| Crystal size (mm) | 0.42 × 0.12 × 0.09 |
| |
| Data collection |
| Diffractometer | Bruker Nonius KappaCCD
diffractometer |
| Absorption correction | Multi-scan
(SADABS; Sheldrick, 2003) |
| Tmin, Tmax | 0.836, 0.961 |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 10725, 1718, 1273 |
| Rint | 0.044 |
| (sin θ/λ)max (Å−1) | 0.650 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.039, 0.097, 1.09 |
| No. of reflections | 1718 |
| No. of parameters | 82 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.28, −0.32 |
Selected geometric parameters (Å, º) top| C2—C3 | 1.503 (2) | C5—Cl1 | 1.7742 (19) |
| C3—C4 | 1.510 (2) | C6—O1 | 1.253 (2) |
| C4—C4i | 1.509 (3) | C6—O2 | 1.236 (2) |
| | | |
| Cl1—C5—C6—O1 | −16.3 (2) | Cl1—C5—C6—O2 | 165.29 (13) |
| Symmetry code: (i) −x+1, −y+1, −z+1. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1A···O1 | 0.88 | 1.95 | 2.801 (2) | 163 |
| N1—H1B···O2ii | 0.88 | 1.86 | 2.739 (2) | 177 |
| N1—H1C···O1iii | 0.88 | 1.99 | 2.814 (2) | 154 |
| N1—H1C···O2iv | 0.88 | 2.59 | 3.020 (2) | 111 |
| Symmetry codes: (ii) −x+1, −y+1, −z; (iii) −x+2, −y+1, −z; (iv) x, y−1, z. |


 journal menu
journal menu organic compounds
organic compounds











![[Figure 1]](https://journals.iucr.org/c/issues/2008/09/00/hj3084/hj3084fig1thm.gif)
![[Figure 2]](https://journals.iucr.org/c/issues/2008/09/00/hj3084/hj3084fig2thm.gif)
![[Figure 3]](https://journals.iucr.org/c/issues/2008/09/00/hj3084/hj3084fig3thm.gif)
![[Figure 4]](https://journals.iucr.org/c/issues/2008/09/00/hj3084/hj3084fig4thm.gif)




A substantial number of salts containing the hexamethylenediammonium cation, [H3N(CH2)6NH3]2+, are recorded in the Cambridge Structural Database (CSD, Version?; Allen 2002), but there are very few entries for unsolvated salts containing simple mono-negative anions which do not contain further potential hydrogen-bond capability. Accordingly, it is of interest to investigate the hydrogen-bonded structure of such a simple salt, and we therefore report here the structure of the title compound, (I) (Fig. 1). Compound (I) was prepared from a reaction between hexamethylenediamine and chloroacetic acid, where the desired product was hexamethylenediglycine intended for use as a flexible linker in the coupling of fullerene units.
In compound (I), the cation lies across a centre of inversion in space group P1, selected for the sake of convenience as that at (1/2, 1/2, 1/2), and it adopts a nearly planar all-trans conformation. The three independent C—C distances within the cation are virtually identical (Table 1). By contrast, in the corresponding chloride salt, where the cation lies in a general position (Borkakoti et al., 1978), there is a marked alternation in the C—C distances between those which are shorter than the mean value and those which are longer, with the distances ranging from 1.507 (4) to 1.538 (4) Å. In the 3,5-dinitrobenzoate salt, the cation lies across a centre of inversion in space group P21/c (Wang & Wei, 2007), and again the C—C distances show a marked alternation of long and short bonds, with distances ranging from 1.499 (5) to 1.539 (3) Å. The saccharinate salt contains two independent cations, both lying across centres of inversion in space group P21/c (Wang et al., 2006). One of the cations exhibits a similar alternation of C—C bond lengths in the range 1.483 (4) to 1.518 (5) Å. The other was refined as disordered, but the reported C—C distances range from 0.970 (10) to 1.571 (6) Å, and no conclusion can be drawn from this apparently inadequate disorder model. These three determinations (Borkakoti et al., 1978; Wang & Wei, 2007; Wang et al., 2006) were all based on diffraction data collected at ambient temperature. The near constancy of the C—C distances found for (I) at 120 K suggests that the bond-length alternation observed in the other salts may possibly be an artefact of the larger thermal motion at ambient temperature.
The two independent C—O distances in the anion are fairly similar, as expected, but the conformation of the anion is such that the C—Cl bond nearly eclipses one of the C—O bonds, as indicated by the relevant torsion angles (Table 1).
The ionic components of (I) are linked into a complex three-dimensional framework structure. Within the selected asymmetric unit (Fig. 1), ammonium atom N1 acts as hydrogen-bond donor, via atom H1A, to carboxylate atom O1. In addition, these aggregates are linked by a second two-centre N—H···O hydrogen bond involving atom H1B and a planar but markedly asymmetric three-centre N—H···(O)2 hydrogen bond involving atom H1C (Table 2). However, the formation of the framework structure is readily analysed in terms of three one-dimensional sub-structures, two of them in the form of rather similar chains of edge-fused rings.
Atom N1 at (x, y, z) acts as hydrogen-bond donor, via atoms H1B and H1C, respectively, to carboxylate atoms O2 in the anion at (1 - x, 1 - y, -z) and O1 in the anion at (2 - x, 1 - y, -z). Propagation of these two hydrogen bonds by translation and inversion then leads to the formation of a chain of edge-fused R44(26) (Bernstein et al., 1995) rings running parallel to the [100] direction in which the rings are centred at (n, 1/2, 1/2), where n represents zero or an integer (Fig. 2). The Cl atoms lie on the exterior of the chains. This structural motif can alternatively be regarded as a molecular ladder, with an anti-parallel pair of C22(6) chains acting as the uprights of the ladder and the cations providing the rungs.
In the longer component of the three-centre interaction, ammonium atom N1 at (x, y, z) acts as hydrogen-bond donor, via atom H1C, to carboxylate atom O2 in the anion at (x, -1 + y, z). Propagation by translation and inversion of this hydrogen bond, together with that within the asymmetric unit, generates a second chain of edge-fused R44(26) rings, this time running parallel to the [010] direction with the rings centred at (1/2, n, 1/2), where n represents zero or an integer (Fig. 3).
In the final sub-structure, ammonium atom N1 at (x, y, z) acts as hydrogen-bond donor to carboxylate atom O1 within the asymmetric unit and to carboxylate atom O2 in the anion at (1 - x, 1 - y, -z), so forming an R44(12) ring centred at (1/2, 1/2, 0). Propagation of this motif by inversion then generates a chain of rings running parallel to the [001] direction with the rings centred at (1/2, 1/2, n), where n represents zero or an integer (Fig. 4). The combination of the chains along [100], [010] and [001] suffices to generate a continuous three-dimensional framework structure. Because each of the sub-structures is itself a chain of rings, the overall three-dimensional structure is of considerable complexity, with many ring motifs embedded within it.
The hydrogen-bonded structure of hexamethylenediammonium bis(3,5-dinitrobenzoate) is only two-dimensional (Wang & Wei, 2007), but three-dimensional hydrogen-bonded structures are present in hexamethylenedammonium di(saccharinate) (Wang et al., 2006), in hexamethylenedammonium dichloride (Borkakoti et al., 1978) and presumably also in the isomorphous dibromide salt (Binnie & Robertson, 1949). The corresponding diiodide salt (CSD refcode HXMAMI; Reference?) has markedly different cell dimensions from the chloride and bromide salts, but no H-atom coordinates are recorded in the CSD and the reported R factor is 0.23. Accordingly, no conclusions can safely be drawn about the hydrogen-bonded structure of this compound.
It is of interest to note that in the title compound, where the anion contains only two potential hydrogen-bond acceptor sites, the overall hydrogen-bonded structure is three-dimensional, whereas in the bis(3,5-dinitrobenzoate) salt (Wang & Wei, 2007), where the anion contains six potential hydrogen-bond acceptor sites, the hydrogen-bonded structure is nonetheless only two-dimensional. In both of these salts, only carboxylate O atoms are involved in the hydrogen bonding, and each structure utilizes all of the N—H bonds, but the overall supramolecular arrangements are very different. This suggests that attempts to make structural predictions for crystal engineering purposes, even for such simple salts, may have only limited success.