The title compound, dimercury(II) divanadium(V) ditellurium(IV) undecaoxide, Hg
2V
2Te
2O
11, is a new representative within the family of divalent oxovanadato(V)tellurates(IV). The anionic framework is made up of disphenoidal [TeO
4] polyhedra that are linked by corner-sharing to two neighbouring pyrovanadate units, resulting in chains of six-membered rings propagating parallel to [1
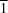
0]. The bridging O atom of the pyrovanadate unit is located on an inversion centre, leading to a staggered conformation and a linear V—O—V angle between the two [VO
4] tetrahedra. The anionic chains are connected by interjacent six-coordinate Hg
2+ cations into a three-dimensional framework. The 5
s2 lone electron pair of the Te
IV atom is stereochemically active and protrudes into the free space of the chain links.
Supporting information
CCDC reference: 1412516
Data collection: SMART (Bruker, 2006); cell refinement: SAINT (Bruker, 2006); data reduction: SAINT (Bruker, 2006); program(s) used to solve structure: SHELXS97 (Sheldrick, 2008); program(s) used to refine structure: SHELXL2014 (Sheldrick, 2015); molecular graphics: ATOMS (Dowty, 2006); software used to prepare material for publication: publCIF (Westrip, 2010).
Dimercury(II) divanadium(V) ditellurium(IV) undecaoxide
top
Crystal data top
| Hg2Te2V2O11 | Z = 1 |
| Mr = 934.26 | F(000) = 398 |
| Triclinic, P1 | Dx = 5.996 Mg m−3 |
| a = 5.4000 (16) Å | Mo Kα radiation, λ = 0.71073 Å |
| b = 5.6093 (17) Å | Cell parameters from 2088 reflections |
| c = 9.132 (3) Å | θ = 3.7–30.9° |
| α = 80.688 (6)° | µ = 36.90 mm−1 |
| β = 72.990 (5)° | T = 293 K |
| γ = 80.080 (5)° | Block, light yellow |
| V = 258.73 (14) Å3 | 0.24 × 0.22 × 0.12 mm |
Data collection top
Bruker SMART APEX CCD
diffractometer | 1515 reflections with I > 2σ(I) |
| Radiation source: fine-focus sealed tube | Rint = 0.022 |
| ω–scans | θmax = 31.0°, θmin = 3.7° |
Absorption correction: multi-scan
(SADABS; Bruker, 2006) | h = −7→7 |
| Tmin = 0.113, Tmax = 0.199 | k = −7→8 |
| 2989 measured reflections | l = −13→13 |
| 1585 independent reflections | |
Refinement top
| Refinement on F2 | Primary atom site location: difference Fourier map |
| Least-squares matrix: full | w = 1/[σ2(Fo2) + (0.0305P)2 + 1.9407P]
where P = (Fo2 + 2Fc2)/3 |
| R[F2 > 2σ(F2)] = 0.027 | (Δ/σ)max = 0.001 |
| wR(F2) = 0.066 | Δρmax = 2.93 e Å−3 |
| S = 1.04 | Δρmin = −2.17 e Å−3 |
| 1585 reflections | Extinction correction: SHELXL2014 (Sheldrick, 2015), Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| 80 parameters | Extinction coefficient: 0.0145 (8) |
| 0 restraints | |
Special details top
Experimental. Face indexing was hampered by poorly developed faces. An absorption correction that 'optimizes' the crystal shape
by using the program
HABITUS (Herrendorf, 1997; University of Giessen, Germany) was attempted.
On basis of the refined
crystal shape (which is always an approximation to the real
crystal form) a
subsequent numerical absorption correction was applied.
The result showed a somewhat worse
Rint (0.04) and slightly higher R1 and wR2 factors
in comparison with the
multi-scan SADABS approach.
For both corrections, the highest remaining electron
densities were virtually the same
(height and distance). |
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes)
are estimated using the full covariance matrix. The cell e.s.d.'s are taken
into account individually in the estimation of e.s.d.'s in distances, angles
and torsion angles; correlations between e.s.d.'s in cell parameters are only
used when they are defined by crystal symmetry. An approximate (isotropic)
treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s.
planes. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| Hg1 | 0.24109 (4) | 0.24323 (4) | 0.97670 (3) | 0.02060 (11) | |
| Te1 | 0.34338 (7) | −0.19514 (7) | 0.73176 (4) | 0.01211 (11) | |
| V1 | −0.14173 (19) | 0.28409 (19) | 0.65183 (12) | 0.0144 (2) | |
| O1 | −0.2740 (11) | 0.1080 (12) | 0.5811 (7) | 0.0325 (12) | |
| O2 | 0.0000 | 0.5000 | 0.5000 | 0.040 (2) | |
| O3 | 0.0888 (9) | 0.1032 (9) | 0.7452 (6) | 0.0209 (9) | |
| O4 | −0.3837 (9) | 0.4327 (9) | 0.7812 (6) | 0.0221 (9) | |
| O5 | 0.4249 (9) | −0.1130 (8) | 0.9002 (5) | 0.0190 (9) | |
| O6 | −0.0847 (8) | 0.3909 (9) | 1.1581 (6) | 0.0200 (9) | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| Hg1 | 0.01871 (14) | 0.01744 (15) | 0.02144 (15) | 0.00835 (9) | −0.00440 (9) | −0.00370 (9) |
| Te1 | 0.01300 (17) | 0.01220 (19) | 0.01056 (17) | 0.00144 (13) | −0.00373 (13) | −0.00199 (13) |
| V1 | 0.0121 (4) | 0.0146 (5) | 0.0123 (4) | 0.0030 (4) | −0.0018 (3) | 0.0026 (4) |
| O1 | 0.028 (2) | 0.043 (3) | 0.029 (3) | −0.003 (2) | −0.011 (2) | −0.010 (3) |
| O2 | 0.039 (4) | 0.031 (4) | 0.029 (4) | 0.003 (4) | 0.007 (4) | 0.016 (3) |
| O3 | 0.020 (2) | 0.018 (2) | 0.024 (2) | 0.0090 (17) | −0.0109 (18) | −0.0039 (18) |
| O4 | 0.0165 (19) | 0.024 (2) | 0.021 (2) | 0.0041 (18) | −0.0018 (17) | −0.0021 (19) |
| O5 | 0.024 (2) | 0.018 (2) | 0.019 (2) | 0.0064 (17) | −0.0151 (18) | −0.0085 (17) |
| O6 | 0.0133 (18) | 0.018 (2) | 0.025 (2) | 0.0000 (16) | 0.0007 (17) | −0.0051 (18) |
Geometric parameters (Å, º) top
| Hg1—O6 | 2.182 (5) | Te1—O6iv | 1.876 (5) |
| Hg1—O5 | 2.197 (4) | Te1—O3 | 1.970 (4) |
| Hg1—O5i | 2.349 (4) | Te1—O4v | 2.400 (5) |
| Hg1—O6ii | 2.374 (5) | V1—O1 | 1.620 (6) |
| Hg1—O4iii | 2.537 (5) | V1—O4 | 1.685 (5) |
| Hg1—O3 | 2.746 (5) | V1—O2 | 1.7692 (11) |
| Hg1—O3iv | 3.217 (5) | V1—O3 | 1.792 (4) |
| Hg1—O4ii | 3.400 (5) | V1—O6ii | 3.296 (5) |
| Te1—O5 | 1.862 (4) | V1—O1vi | 3.379 (6) |
| | | |
| O6—Hg1—O5 | 138.66 (19) | O6ii—V1—O1vi | 120.07 (12) |
| O6—Hg1—O5i | 103.38 (17) | V1vii—O2—V1 | 180.0 |
| O5—Hg1—O5i | 74.30 (17) | V1—O3—Te1 | 142.3 (3) |
| O6—Hg1—O6ii | 78.7 (2) | V1—O3—Hg1 | 124.5 (2) |
| O5—Hg1—O6ii | 131.49 (17) | Te1—O3—Hg1 | 92.76 (16) |
| O5i—Hg1—O6ii | 136.92 (17) | V1—O3—Hg1iv | 106.2 (2) |
| O6—Hg1—O4iii | 132.33 (18) | Te1—O3—Hg1iv | 81.09 (16) |
| O5—Hg1—O4iii | 88.53 (18) | Hg1—O3—Hg1iv | 83.30 (13) |
| O5i—Hg1—O4iii | 79.60 (17) | V1—O4—Te1viii | 127.3 (3) |
| O6ii—Hg1—O4iii | 69.22 (15) | V1—O4—Hg1ix | 125.4 (3) |
| O6—Hg1—O3 | 113.52 (16) | Te1viii—O4—Hg1ix | 93.00 (15) |
| O5—Hg1—O3 | 63.37 (14) | V1—O4—Hg1ii | 120.3 (2) |
| O5i—Hg1—O3 | 136.85 (14) | Te1viii—O4—Hg1ii | 82.72 (14) |
| O6ii—Hg1—O3 | 74.12 (16) | Hg1ix—O4—Hg1ii | 97.23 (15) |
| O4iii—Hg1—O3 | 90.92 (16) | V1—O4—Hg1iv | 76.76 (17) |
| O6—Hg1—O3iv | 58.05 (15) | Te1viii—O4—Hg1iv | 152.87 (17) |
| O5—Hg1—O3iv | 80.87 (16) | Hg1ix—O4—Hg1iv | 60.22 (10) |
| O5i—Hg1—O3iv | 84.05 (15) | Hg1ii—O4—Hg1iv | 95.65 (11) |
| O6ii—Hg1—O3iv | 128.12 (13) | Te1—O5—Hg1 | 116.5 (2) |
| O4iii—Hg1—O3iv | 162.44 (13) | Te1—O5—Hg1i | 135.7 (2) |
| O3—Hg1—O3iv | 96.70 (13) | Hg1—O5—Hg1i | 105.70 (17) |
| O6—Hg1—O4ii | 62.79 (15) | Te1—O5—V1iii | 90.59 (18) |
| O5—Hg1—O4ii | 129.99 (13) | Hg1—O5—V1iii | 78.01 (14) |
| O5i—Hg1—O4ii | 55.69 (13) | Hg1i—O5—V1iii | 85.28 (14) |
| O6ii—Hg1—O4ii | 90.62 (15) | Te1—O5—Hg1iv | 71.75 (14) |
| O4iii—Hg1—O4ii | 82.77 (15) | Hg1—O5—Hg1iv | 82.74 (13) |
| O3—Hg1—O4ii | 164.72 (13) | Hg1i—O5—Hg1iv | 128.90 (18) |
| O3iv—Hg1—O4ii | 93.32 (12) | V1iii—O5—Hg1iv | 144.51 (13) |
| O5—Te1—O6iv | 97.7 (2) | Te1—O5—Hg1x | 68.66 (14) |
| O5—Te1—O3 | 86.95 (19) | Hg1—O5—Hg1x | 136.30 (19) |
| O6iv—Te1—O3 | 92.2 (2) | Hg1i—O5—Hg1x | 90.39 (14) |
| O5—Te1—O4v | 82.59 (19) | V1iii—O5—Hg1x | 144.81 (14) |
| O6iv—Te1—O4v | 80.53 (18) | Hg1iv—O5—Hg1x | 56.66 (7) |
| O3—Te1—O4v | 166.31 (19) | Te1—O5—V1iv | 131.7 (2) |
| O5—Te1—Hg1 | 34.69 (14) | Hg1—O5—V1iv | 77.96 (13) |
| O1—V1—O4 | 107.3 (3) | Hg1i—O5—V1iv | 68.26 (11) |
| O1—V1—O2 | 108.0 (2) | V1iii—O5—V1iv | 137.41 (12) |
| O4—V1—O2 | 108.63 (19) | Hg1iv—O5—V1iv | 64.52 (8) |
| O1—V1—O3 | 109.5 (3) | Hg1x—O5—V1iv | 70.75 (9) |
| O4—V1—O3 | 110.0 (2) | Te1iv—O6—Hg1 | 119.1 (2) |
| O2—V1—O3 | 113.25 (18) | Te1iv—O6—Hg1ii | 114.4 (2) |
| O1—V1—O6ii | 172.3 (2) | Hg1—O6—Hg1ii | 101.3 (2) |
| O4—V1—O6ii | 67.99 (19) | Te1iv—O6—V1ii | 119.0 (2) |
| O2—V1—O6ii | 79.56 (10) | Hg1—O6—V1ii | 108.47 (16) |
| O3—V1—O6ii | 67.79 (18) | Hg1ii—O6—V1ii | 89.41 (14) |
| O1—V1—O1vi | 63.7 (3) | Te1iv—O6—V1iv | 61.66 (13) |
| O4—V1—O1vi | 168.6 (2) | Hg1—O6—V1iv | 75.75 (13) |
| O2—V1—O1vi | 81.61 (12) | Hg1ii—O6—V1iv | 171.44 (19) |
| O3—V1—O1vi | 68.90 (18) | V1ii—O6—V1iv | 99.15 (12) |
| Symmetry codes: (i) −x+1, −y, −z+2; (ii) −x, −y+1, −z+2; (iii) x+1, y, z; (iv) −x, −y, −z+2; (v) x+1, y−1, z; (vi) −x, −y, −z+1; (vii) −x, −y+1, −z+1; (viii) x−1, y+1, z; (ix) x−1, y, z; (x) x, y−1, z. |
Bond-valence-sum calculations in valence units top| Atom (expected valence state) | O1 (2) | O2 (2) | O3 (2) | O4 (2) | O5 (2) | O6 (2) | Σ | Δ to expected |
| Hg1 (2) | | | 0.123 | 0.217 | 0.544, 0.361 | 0.567, 0.337 | 2.149 | 0.149 |
| Te1 (4) | | | 1.019 | 0.319 | 1.365 | 1.314 | 4.016 | 0.016 |
| V1 (5) | 1.644 | 1.096 (2O) | 1.030 | 1.376 | | | 5.146 | 0.146 |
| Σ | 1.644 | 2.192 | 2.172 | 1.912 | 2.270 | 2.218 | | |
| Δ to expected | 0.356 | 0.192 | 0.172 | 0.088 | 0.270 | 0.218 | | |
| Calculated with the parameters compiled by Brese & O'Keeffe (1991). |


 journal menu
journal menu research papers
research papers














