The title compound, C
7H
9O
4P, obtained as a by-product of the reaction between Lawesson's reagent, (I), and CH
3I, can be recognized as the final product of the S/O interchange reaction at the P atom of (I). Hydrogen bonds of type P—O—H

O=P link molecules into helical chains and form ten-membered hydrogen-bonded rings with the graph-set notation
R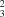
(10). Weaker intermolecular contacts between P—O and a phenyl H atom link the chains into a three-dimensional lattice. The parent benzenephosphonic acid [Weakley (1976).
Acta Cryst. B
32, 2889–2890] does not adopt an analogous structure, but its arsenic analogue [Shimada (1960).
Bull. Chem. Soc. Jpn,
33, 301–304] does and can be regarded as isostructural. We rationalize these three structures in terms of their significant intermolecular interactions.
Supporting information
CCDC reference: 187925
Lawesson's reagent, (I) (0.50 g, 1.2 mmol), was dissolved in toluene (60 ml,
dried over molecular sieves) under a dinitrogen atmosphere, and then a
fourfold stoichiometric excess of CH3I was added. The reaction mixture was
refluxed for 5 h and the resulting solid was then filtered off. Compound (II)
separated from the filtrate as white needles after two weeks standing in the
open atmosphere. Elemental analysis (%), found (calculated for C7H9O4P):
C 44.53 (44.69), H 4.71 (4.82).
A meaningful Flack parameter (Flack, 1983) could not be determined; it refined
to a value of 0.4 (2) and so Friedel opposites were merged for the final stages
of the refinement. Hydroxyl and methyl H atoms were located from ΔF syntheses
and thereafter refined as part of rigid rotating groups, with Uiso(H)
= 1.5Ueq(O,C); aromatic H atoms were placed geometrically and
then allowed to refine riding on their parent atoms, with Uiso(H) =
1.2Ueq(C). The following distance constraints were applied: O—H =
0.84, C—H(methyl) = 0.98 and C—H(aromatic) = 0.95 Å.
Data collection: SMART (Bruker, 1998); cell refinement: SAINT (Bruker, 1998); data reduction: SAINT and SHELXTL (Bruker, 1997); program(s) used to solve structure: SHELXS97 (Sheldrick, 1990); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics: SHELXTL; software used to prepare material for publication: SHELXL97 and PLATON (Spek, 2001).
Crystal data top
| C7H9O4P | Dx = 1.495 Mg m−3 |
| Mr = 188.11 | Mo Kα radiation, λ = 0.71073 Å |
| Orthorhombic, P212121 | Cell parameters from 1415 reflections |
| a = 4.5819 (6) Å | θ = 2.3–26.4° |
| b = 10.0867 (13) Å | µ = 0.30 mm−1 |
| c = 18.087 (2) Å | T = 150 K |
| V = 835.91 (18) Å3 | Lath, colourless |
| Z = 4 | 0.31 × 0.07 × 0.04 mm |
| F(000) = 392 | |
Data collection top
Bruker SMART1000 CCD area-detector
diffractometer | 939 reflections with I > 2σ(I) |
| Radiation source: normal-focus sealed tube | Rint = 0.090 |
| Graphite monochromator | θmax = 28.8°, θmin = 2.3° |
| ω scans | h = −5→6 |
| 5423 measured reflections | k = −11→13 |
| 1210 independent reflections | l = −24→22 |
Refinement top
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Hydrogen site location: see text |
| R[F2 > 2σ(F2)] = 0.042 | H-atom parameters constrained |
| wR(F2) = 0.094 | w = 1/[σ2(Fo2) + (0.05P)2]
where P = (Fo2 + 2Fc2)/3 |
| S = 0.94 | (Δ/σ)max = 0.001 |
| 1210 reflections | Δρmax = 0.30 e Å−3 |
| 112 parameters | Δρmin = −0.31 e Å−3 |
| 0 restraints | |
Crystal data top
| C7H9O4P | V = 835.91 (18) Å3 |
| Mr = 188.11 | Z = 4 |
| Orthorhombic, P212121 | Mo Kα radiation |
| a = 4.5819 (6) Å | µ = 0.30 mm−1 |
| b = 10.0867 (13) Å | T = 150 K |
| c = 18.087 (2) Å | 0.31 × 0.07 × 0.04 mm |
Data collection top
Bruker SMART1000 CCD area-detector
diffractometer | 939 reflections with I > 2σ(I) |
| 5423 measured reflections | Rint = 0.090 |
| 1210 independent reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.042 | 0 restraints |
| wR(F2) = 0.094 | H-atom parameters constrained |
| S = 0.94 | Δρmax = 0.30 e Å−3 |
| 1210 reflections | Δρmin = −0.31 e Å−3 |
| 112 parameters | |
Special details top
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes)
are estimated using the full covariance matrix. The cell e.s.d.'s are taken
into account individually in the estimation of e.s.d.'s in distances, angles
and torsion angles; correlations between e.s.d.'s in cell parameters are only
used when they are defined by crystal symmetry. An approximate (isotropic)
treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s.
planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor
wR and goodness of fit S are based on F2, conventional
R-factors R are based on F, with F set to zero for
negative F2. The threshold expression of F2 >
σ(F2) is used only for calculating R-factors(gt) etc.
and is not relevant to the choice of reflections for refinement.
R-factors based on F2 are statistically about twice as large
as those based on F, and R- factors based on ALL data will be
even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| P | 0.09220 (18) | 0.68568 (9) | 0.89583 (4) | 0.0233 (2) | |
| O1 | 0.3532 (5) | 0.6510 (2) | 0.94311 (10) | 0.0296 (6) | |
| O2 | −0.0425 (5) | 0.8220 (2) | 0.91755 (11) | 0.0296 (6) | |
| H2O | −0.0879 | 0.8211 | 0.9625 | 0.044* | |
| O3 | −0.1416 (5) | 0.5749 (2) | 0.90187 (13) | 0.0293 (5) | |
| H3O | −0.3037 | 0.6089 | 0.9125 | 0.044* | |
| C1 | 0.1925 (7) | 0.6997 (3) | 0.80158 (16) | 0.0232 (7) | |
| C2 | 0.0942 (8) | 0.6082 (3) | 0.74983 (16) | 0.0289 (7) | |
| H2 | −0.0402 | 0.5415 | 0.7645 | 0.035* | |
| C3 | 0.1895 (7) | 0.6131 (3) | 0.67714 (17) | 0.0299 (8) | |
| H3 | 0.1227 | 0.5494 | 0.6423 | 0.036* | |
| C4 | 0.3823 (8) | 0.7111 (3) | 0.65571 (16) | 0.0302 (8) | |
| C5 | 0.4766 (8) | 0.8056 (4) | 0.70600 (17) | 0.0331 (8) | |
| H5 | 0.6043 | 0.8744 | 0.6906 | 0.040* | |
| C6 | 0.3847 (8) | 0.7994 (3) | 0.77833 (16) | 0.0289 (7) | |
| H6 | 0.4524 | 0.8633 | 0.8129 | 0.035* | |
| O4 | 0.4929 (6) | 0.7233 (2) | 0.58565 (12) | 0.0422 (7) | |
| C4M | 0.4369 (11) | 0.6186 (4) | 0.53464 (18) | 0.0474 (11) | |
| H4M1 | 0.5051 | 0.5345 | 0.5556 | 0.071* | |
| H4M2 | 0.5403 | 0.6361 | 0.4882 | 0.071* | |
| H4M3 | 0.2267 | 0.6131 | 0.5250 | 0.071* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| P | 0.0189 (4) | 0.0278 (4) | 0.0233 (4) | 0.0005 (4) | 0.0001 (4) | 0.0006 (4) |
| O1 | 0.0193 (12) | 0.0454 (15) | 0.0240 (11) | 0.0022 (10) | −0.0021 (9) | 0.0016 (10) |
| O2 | 0.0314 (13) | 0.0345 (13) | 0.0227 (10) | 0.0034 (12) | 0.0042 (9) | −0.0028 (10) |
| O3 | 0.0182 (12) | 0.0324 (12) | 0.0374 (12) | 0.0004 (10) | 0.0026 (11) | 0.0027 (12) |
| C1 | 0.0181 (15) | 0.0265 (17) | 0.0251 (14) | 0.0050 (15) | −0.0017 (12) | −0.0010 (15) |
| C2 | 0.0246 (17) | 0.0311 (18) | 0.0310 (17) | −0.0025 (17) | −0.0012 (16) | −0.0011 (14) |
| C3 | 0.0347 (19) | 0.0303 (19) | 0.0246 (16) | 0.0002 (17) | −0.0046 (14) | −0.0068 (15) |
| C4 | 0.0367 (19) | 0.0341 (19) | 0.0197 (14) | 0.0073 (17) | −0.0009 (14) | 0.0049 (14) |
| C5 | 0.040 (2) | 0.0312 (18) | 0.0275 (16) | −0.0066 (18) | −0.0008 (14) | 0.0049 (16) |
| C6 | 0.0317 (18) | 0.0309 (18) | 0.0241 (15) | −0.0037 (19) | −0.0021 (14) | −0.0046 (14) |
| O4 | 0.0628 (18) | 0.0394 (15) | 0.0242 (12) | −0.0062 (13) | 0.0088 (11) | −0.0036 (10) |
| C4M | 0.076 (3) | 0.042 (2) | 0.0245 (16) | −0.001 (2) | 0.009 (2) | −0.0067 (16) |
Geometric parameters (Å, º) top
| P—O1 | 1.511 (2) | C3—H3 | 0.95 |
| P—O2 | 1.557 (2) | C4—O4 | 1.370 (4) |
| P—O3 | 1.552 (2) | C4—C5 | 1.387 (5) |
| P—C1 | 1.771 (3) | C5—C6 | 1.376 (4) |
| O2—H2O | 0.84 | C5—H5 | 0.95 |
| O3—H3O | 0.84 | C6—H6 | 0.95 |
| C1—C2 | 1.390 (4) | O4—C4M | 1.426 (4) |
| C1—C6 | 1.401 (5) | C4M—H4M1 | 0.98 |
| C2—C3 | 1.386 (4) | C4M—H4M2 | 0.98 |
| C2—H2 | 0.95 | C4M—H4M3 | 0.98 |
| C3—C4 | 1.382 (5) | | |
| | | |
| O1—P—O2 | 112.03 (13) | O4—C4—C3 | 124.1 (3) |
| O1—P—O3 | 109.88 (13) | O4—C4—C5 | 115.4 (3) |
| O2—P—O3 | 110.12 (12) | C3—C4—C5 | 120.5 (3) |
| O1—P—C1 | 110.97 (14) | C6—C5—C4 | 119.8 (3) |
| O2—P—C1 | 105.96 (14) | C6—C5—H5 | 120.1 |
| O3—P—C1 | 107.73 (15) | C4—C5—H5 | 120.1 |
| P—O2—H2O | 109.5 | C5—C6—C1 | 120.7 (3) |
| P—O3—H3O | 109.5 | C5—C6—H6 | 119.6 |
| C2—C1—C6 | 118.6 (3) | C1—C6—H6 | 119.6 |
| C2—C1—P | 120.7 (3) | C4—O4—C4M | 117.7 (3) |
| C6—C1—P | 120.6 (2) | O4—C4M—H4M1 | 109.5 |
| C3—C2—C1 | 120.9 (3) | O4—C4M—H4M2 | 109.5 |
| C3—C2—H2 | 119.6 | H4M1—C4M—H4M2 | 109.5 |
| C1—C2—H2 | 119.6 | O4—C4M—H4M3 | 109.5 |
| C4—C3—C2 | 119.5 (3) | H4M1—C4M—H4M3 | 109.5 |
| C4—C3—H3 | 120.2 | H4M2—C4M—H4M3 | 109.5 |
| C2—C3—H3 | 120.2 | | |
| | | |
| O1—P—C1—C2 | 112.0 (3) | C2—C3—C4—O4 | 179.0 (3) |
| O3—P—C1—C2 | −8.3 (3) | C2—C3—C4—C5 | −1.1 (5) |
| O2—P—C1—C2 | −126.1 (3) | O4—C4—C5—C6 | −178.0 (3) |
| O1—P—C1—C6 | −64.7 (3) | C3—C4—C5—C6 | 2.0 (5) |
| O3—P—C1—C6 | 175.0 (2) | C4—C5—C6—C1 | −1.1 (5) |
| O2—P—C1—C6 | 57.2 (3) | C2—C1—C6—C5 | −0.7 (5) |
| C6—C1—C2—C3 | 1.6 (5) | P—C1—C6—C5 | 176.1 (3) |
| P—C1—C2—C3 | −175.1 (3) | C3—C4—O4—C4M | −8.8 (5) |
| C1—C2—C3—C4 | −0.7 (5) | C5—C4—O4—C4M | 171.2 (3) |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| O2—H2O···O1i | 0.84 | 1.75 | 2.580 (3) | 169 |
| O3—H3O···O1ii | 0.84 | 1.72 | 2.550 (3) | 169 |
| C3—H3···O2iii | 0.95 | 2.56 | 3.465 (4) | 159 |
| Symmetry codes: (i) x−1/2, −y+3/2, −z+2; (ii) x−1, y, z; (iii) −x, y−1/2, −z+3/2. |
Experimental details
| Crystal data |
| Chemical formula | C7H9O4P |
| Mr | 188.11 |
| Crystal system, space group | Orthorhombic, P212121 |
| Temperature (K) | 150 |
| a, b, c (Å) | 4.5819 (6), 10.0867 (13), 18.087 (2) |
| V (Å3) | 835.91 (18) |
| Z | 4 |
| Radiation type | Mo Kα |
| µ (mm−1) | 0.30 |
| Crystal size (mm) | 0.31 × 0.07 × 0.04 |
| |
| Data collection |
| Diffractometer | Bruker SMART1000 CCD area-detector
diffractometer |
| Absorption correction | – |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 5423, 1210, 939 |
| Rint | 0.090 |
| (sin θ/λ)max (Å−1) | 0.678 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.042, 0.094, 0.94 |
| No. of reflections | 1210 |
| No. of parameters | 112 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.30, −0.31 |
Selected geometric parameters (Å, º) top| P—O1 | 1.511 (2) | P—O3 | 1.552 (2) |
| P—O2 | 1.557 (2) | P—C1 | 1.771 (3) |
| | | |
| O1—P—O2 | 112.03 (13) | O1—P—C1 | 110.97 (14) |
| O1—P—O3 | 109.88 (13) | O2—P—C1 | 105.96 (14) |
| O2—P—O3 | 110.12 (12) | O3—P—C1 | 107.73 (15) |
| | | |
| C3—C4—O4—C4M | −8.8 (5) | C5—C4—O4—C4M | 171.2 (3) |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| O2—H2O···O1i | 0.84 | 1.75 | 2.580 (3) | 169 |
| O3—H3O···O1ii | 0.84 | 1.72 | 2.550 (3) | 169 |
| Symmetry codes: (i) x−1/2, −y+3/2, −z+2; (ii) x−1, y, z. |


 journal menu
journal menu organic compounds
organic compounds











![[Figure 1]](http://journals.iucr.org/c/issues/2002/05/00/na1560/na1560fig1thm.gif)
![[Figure 2]](http://journals.iucr.org/c/issues/2002/05/00/na1560/na1560fig2thm.gif)




It is widely recognized that 2,4-bis(4-methoxyphenyl)-2,4-dithio-1,3,2,4-dithiadiphosphetane [Lawesson's reagent, (I); Yde et al., 1984; Scheibye et al., 1982; Shabana et al., 1980; Scheibye et al., 1978a,b; Pedersen et al., 1978] is among the most effective thionating agents for organic molecules and plays a crucial role in the synthesis of phosphorus-containing rings (Cherkasov et al., 1985; Foreman & Woolins, 2000). The wide range of applications, along with the variety of transformations and new products that can be made using it, make it one of the most widely used and versatile reagents. Recently, while exploring the high reactivity of (I) towards nucleophilic reagents, which is one of its most characteristic features, we obtained new phosphonodithioates and amidophosphonodithioates and their metal complexes (Arca et al., 1997; Aragoni et al., 2000).
In an attempt to explore further the reactivity of Lawesson's reagent, 4-methoxyphenylphosphonic acid, (II), has been obtained as a by-product from the reaction of (I) with methyl iodide. Interestingly, a similar reaction between a structural analogue of (I) (i.e. 2,4-diphenyl-2,4-dithio-1,3,2,4-dithiadiphosphetane) and methyl bromide in a sealed tube gives methyl phenylphosphonobromidodithioate in almost quantitative yield (Von Fluk & Binder, 1967). Compound (II) can be considered the final desulfurated product in the S/O interchange reaction taking place at the P atom of (I) during thionation reactions. In this case, the O atoms probably come from traces of water in the solvent and/or from air during crystallization. A compound similar to (II) [structure (III) in the Scheme above] was obtained from the reaction of 2,4-(naphthalene-1,8-diyl)-2,4-dithio-1,3,2,4-dithiadiphosphetane with ethylene glycol at 413 K (Kilian et al., 1999). As in the structure of (II), the two P atoms are coordinated to four O atoms which come from ethylene glycol, but a P—O—P bridge is built into a six-membered C3P2O heterocycle due to the rigidity of the organyl backbone.
The intramolecular geometry of (II) is unexceptional and we note only the distinction between the two equivalent P—O single bonds of 1.552 (2) and 1.557 (2) Å and the P═O distance of 1.511 (2) Å (Fig. 1 and Table 1), and the typical coplanarity of the anisole methoxy substituent at C4 with its phenyl ring [e.g. C3—C4—O4—C4M -8.8 (5)°]. However, there are two unique but geometrically similar hydrogen bonds of type P—O—H···O═P (Fig. 2 and Table 2), which result in the formation of ten-membered hydrogen-bonded rings, each containing three P—OH donors and two P═O acceptors, giving a graph-set notation R23(10) (Bernstein et al., 1995). Successive molecules are related by the operation of a 21 screw axis parallel to a. The overall helical structure along a is generated by repetition of these rings in that direction. In addition, there is a weak interaction between an H atom on the phenyl ring and the P—O related by the screw axis parallel to b, which links the chains into a three-dimensional lattice [C3—H3···O2(-x,-1/2 + y,3/2 - z) H···O 2.56 Å, C···O 3.465 (4) Å and C—H···O 159°].
There are very few structures of arylphosphonic acids in the literature: in fact, we have identified only three, two of which (Nieger et al., 1999a,b) are dimethyl sulphoxide solvates where the solvent molecules are involved in the hydrogen bonding, and which are therefore not comparable with the title compound. The only immediate comparison is with the parent benzenephosphonic acid PhP(O)(OH)2 (Weakley, 1976) in which hydrogen bonding produces puckered layers of molecules with only weak contacts between these layers. Each P—OH group participates in one hydrogen bond but the O atom of the P═O group accepts two hydrogen bonds. In contrast to the title compound, the layers in PhP(O)(OH)2 contain centrosymmetric pairs of molecules. The chains observed in the title compound are, therefore, more analogous to the structure of PhAs(O)(OH)2 (Shimada, 1960) than to that of PhP(O)(OH)2 (Weakley, 1976). Allowing for an extension of the c axis caused by the addition of the 4-methoxy substituent, the unit-cell dimensions are comparable [a = 4.70, b = 10.42, c = 14.92 Å for PhAs(O)(OH)2] and the compounds adopt the same space group, P212121, and the same packing motif via hydrogen bonding. They can, therefore, be regarded as isostructural.
We have attempted to rationalize the relationship of the structures of (II), PhAs(O)(OH)2 and PhP(O)(OH)2. In the last of these, any extension to the length of the molecule, whether by substitution at the 4-position of the phenyl ring or by exchanging As for P, would push the layers further apart, thereby destabilizing the structure. In contrast, the motif assumed by (II) and PhAs(O)(OH)2 depends on strong hydrogen bonding along the a direction and weaker interactions along b, but is rather tolerant of extension in the c direction as there are no important interactions along that axis. It would be interesting to explore the limits of this tolerance by increasing the length of the substituent on C4.