Two structural isomers, 3,6-bis(2-chlorophenyl)-1,4-dihydro-1,2,4,5-tetrazine, (I), and 3,5-bis(2-chlorophenyl)-4-amino-1
H-1,2,4-triazole, (II), both C
14H
10Cl
2N
4, form chain-like structures in the solid state, stabilized by N-H

N and N-H
Cl hydrogen bonds. A contribution from weak interactions to the strong hydrogen-bond network is observed in both structures. The secondary graph sets for intermolecular hydrogen bonds [
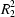
(11) for (I) and
(12) for (II)] indicate the similarity between the networks.
Supporting information
CCDC references: 231074; 231075
Compound (I) was prepared using a modification of the procedure described by Brooker et al. (1987), by reacting bis-(α,2-dichlorobenzilidene)hydrazine and hydrazine hydrate at low temperature. Crystals suitable for X-ray diffraction were crystallized from ethanol, precipitating in the form of yellow–orange needles (m.p. 482–483 K). Compound (I) was dissolved in an acidic water solution and heated to above 353 K, giving the structural isomer (II). The isomerization reaction is irreversible. Well shaped crystals of (II), in the form of colourless prisms (m.p. 441–442 K), were obtained from ethanol. Detailed descriptions of the products and synthesis routes are given elsewhere (Włostowski & Olszewski, 2003).
Since the absorption coefficients of both compounds were comparatively high, a numerical correction (Gaussian integration) based on a well defined crystal shape was applied using SHELX76 (Sheldrick, 1976). For both compounds, H atoms were found from a difference Fourier map and their positional and isotropic displacement parameters were refined.
For both compounds, data collection: P3/P4-PC Software (Siemens, 1991). Cell refinement: P3/P4-PC Software for (I); P3/P4-PC for (II). For both compounds, data reduction: XDISK (Siemens, 1991); program(s) used to solve structure: SHELXS86 (Sheldrick, 1990); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics: ORTEPIII (Burnett & Johnson, 1996); software used to prepare material for publication: SHELXL97 and PLATON (Spek, 2003).
(I) 3,6-bis(2-chlorophenyl)-1,4-dihydro-1,2,4,5-tetrazine
top
Crystal data top
| C14H10Cl2N4 | F(000) = 624 |
| Mr = 305.16 | Dx = 1.495 Mg m−3 |
| Monoclinic, C2/c | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -C 2yc | Cell parameters from 30 reflections |
| a = 16.0020 (12) Å | θ = 7.7–16.4° |
| b = 9.4123 (8) Å | µ = 0.47 mm−1 |
| c = 10.5752 (7) Å | T = 293 K |
| β = 121.664 (5)° | Needle, yellow–orange |
| V = 1355.69 (18) Å3 | 0.62 × 0.09 × 0.09 mm |
| Z = 4 | |
Data collection top
Siemens, P3
diffractometer | 996 reflections with I > 2σ(I) |
| Radiation source: fine-focus sealed tube | Rint = 0.014 |
| Graphite monochromator | θmax = 25.0°, θmin = 2.6° |
| profile data from ω–2θ scans | h = −18→18 |
Absorption correction: gaussian
SHELX76 (Sheldrick, 1976) | k = −11→11 |
| Tmin = 0.936, Tmax = 0.960 | l = −12→12 |
| 2496 measured reflections | 1 standard reflections every 70 reflections |
| 1203 independent reflections | intensity decay: 1.8% |
Refinement top
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.036 | All H-atom parameters refined |
| wR(F2) = 0.099 | w = 1/[σ2(Fo2) + (0.0452P)2 + 1.4393P]
where P = (Fo2 + 2Fc2)/3 |
| S = 1.04 | (Δ/σ)max < 0.001 |
| 1203 reflections | Δρmax = 0.39 e Å−3 |
| 112 parameters | Δρmin = −0.43 e Å−3 |
| 0 restraints | Extinction correction: SHELXL97, Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| Primary atom site location: structure-invariant direct methods | Extinction coefficient: 0.0047 (12) |
Crystal data top
| C14H10Cl2N4 | V = 1355.69 (18) Å3 |
| Mr = 305.16 | Z = 4 |
| Monoclinic, C2/c | Mo Kα radiation |
| a = 16.0020 (12) Å | µ = 0.47 mm−1 |
| b = 9.4123 (8) Å | T = 293 K |
| c = 10.5752 (7) Å | 0.62 × 0.09 × 0.09 mm |
| β = 121.664 (5)° | |
Data collection top
Siemens, P3
diffractometer | 996 reflections with I > 2σ(I) |
Absorption correction: gaussian
SHELX76 (Sheldrick, 1976) | Rint = 0.014 |
| Tmin = 0.936, Tmax = 0.960 | 1 standard reflections every 70 reflections |
| 2496 measured reflections | intensity decay: 1.8% |
| 1203 independent reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.036 | 0 restraints |
| wR(F2) = 0.099 | All H-atom parameters refined |
| S = 1.04 | Δρmax = 0.39 e Å−3 |
| 1203 reflections | Δρmin = −0.43 e Å−3 |
| 112 parameters | |
Special details top
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| Cl1 | 0.16401 (5) | 0.74870 (6) | 0.21918 (8) | 0.0646 (3) | |
| N1 | 0.04530 (12) | 0.4499 (2) | 0.18150 (18) | 0.0376 (4) | |
| N2 | 0.05456 (12) | 0.49908 (19) | 0.40415 (17) | 0.0372 (4) | |
| C1 | 0.20247 (14) | 0.5248 (2) | 0.4027 (2) | 0.0359 (5) | |
| C2 | 0.24122 (16) | 0.6321 (2) | 0.3587 (2) | 0.0439 (5) | |
| C3 | 0.3429 (2) | 0.6493 (3) | 0.4296 (3) | 0.0618 (8) | |
| C4 | 0.4044 (2) | 0.5597 (4) | 0.5428 (3) | 0.0646 (8) | |
| C5 | 0.36748 (18) | 0.4549 (3) | 0.5887 (3) | 0.0589 (7) | |
| C6 | 0.26745 (15) | 0.4384 (3) | 0.5202 (2) | 0.0434 (5) | |
| C10 | 0.09617 (14) | 0.4949 (2) | 0.3295 (2) | 0.0334 (5) | |
| H1 | 0.0749 (15) | 0.470 (2) | 0.134 (2) | 0.036 (5)* | |
| H3 | 0.362 (2) | 0.720 (3) | 0.398 (3) | 0.072 (9)* | |
| H4 | 0.475 (2) | 0.568 (4) | 0.593 (3) | 0.094 (10)* | |
| H5 | 0.412 (2) | 0.391 (3) | 0.670 (3) | 0.078 (9)* | |
| H6 | 0.2407 (16) | 0.363 (3) | 0.552 (2) | 0.049 (6)* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| Cl1 | 0.0958 (6) | 0.0467 (4) | 0.0745 (5) | 0.0089 (3) | 0.0608 (4) | 0.0122 (3) |
| N1 | 0.0339 (9) | 0.0530 (11) | 0.0309 (9) | 0.0034 (8) | 0.0204 (8) | −0.0022 (8) |
| N2 | 0.0340 (9) | 0.0513 (10) | 0.0298 (8) | −0.0032 (7) | 0.0191 (7) | 0.0000 (7) |
| C1 | 0.0388 (11) | 0.0407 (11) | 0.0350 (10) | −0.0041 (9) | 0.0242 (9) | −0.0080 (8) |
| C2 | 0.0558 (13) | 0.0434 (12) | 0.0480 (12) | −0.0080 (10) | 0.0380 (11) | −0.0100 (10) |
| C3 | 0.0720 (18) | 0.0631 (16) | 0.0756 (18) | −0.0309 (15) | 0.0560 (16) | −0.0241 (15) |
| C4 | 0.0409 (14) | 0.091 (2) | 0.0577 (16) | −0.0135 (14) | 0.0232 (13) | −0.0206 (15) |
| C5 | 0.0424 (13) | 0.0792 (19) | 0.0461 (13) | −0.0030 (13) | 0.0172 (11) | −0.0095 (13) |
| C6 | 0.0378 (12) | 0.0531 (14) | 0.0372 (11) | −0.0024 (10) | 0.0181 (10) | −0.0040 (10) |
| C10 | 0.0370 (11) | 0.0366 (10) | 0.0309 (10) | −0.0004 (8) | 0.0208 (9) | 0.0004 (8) |
Geometric parameters (Å, º) top
| Cl1—C2 | 1.730 (2) | C2—C3 | 1.399 (3) |
| N1—C10 | 1.398 (2) | C3—C4 | 1.369 (4) |
| N1—N2i | 1.438 (2) | C3—H3 | 0.87 (3) |
| N1—H1 | 0.88 (2) | C4—C5 | 1.363 (4) |
| N2—C10 | 1.273 (2) | C4—H4 | 0.97 (3) |
| N2—N1i | 1.438 (2) | C5—C6 | 1.376 (3) |
| C10—C1 | 1.480 (3) | C5—H5 | 0.98 (3) |
| C1—C2 | 1.388 (3) | C6—H6 | 0.98 (2) |
| C1—C6 | 1.389 (3) | | |
| | | |
| C10—N1—N2i | 114.14 (15) | C4—C3—C2 | 119.8 (3) |
| C10—N1—H1 | 114.1 (14) | C4—C3—H3 | 125 (2) |
| N2i—N1—H1 | 109.0 (13) | C2—C3—H3 | 115 (2) |
| C10—N2—N1i | 111.68 (16) | C5—C4—C3 | 120.7 (2) |
| N2—C10—N1 | 121.13 (18) | C5—C4—H4 | 117 (2) |
| N2—C10—C1 | 120.07 (17) | C3—C4—H4 | 122 (2) |
| N1—C10—C1 | 118.55 (16) | C4—C5—C6 | 119.7 (3) |
| C2—C1—C6 | 118.02 (19) | C4—C5—H5 | 120.6 (17) |
| C2—C1—C10 | 123.78 (19) | C6—C5—H5 | 119.7 (17) |
| C6—C1—C10 | 118.17 (18) | C5—C6—C1 | 121.5 (2) |
| C1—C2—C3 | 120.2 (2) | C5—C6—H6 | 120.0 (13) |
| C1—C2—Cl1 | 120.20 (17) | C1—C6—H6 | 118.4 (13) |
| C3—C2—Cl1 | 119.56 (19) | | |
| | | |
| N1i—N2—C10—N1 | 1.4 (3) | C6—C1—C2—Cl1 | −176.82 (16) |
| N1i—N2—C10—C1 | −172.72 (17) | C10—C1—C2—Cl1 | 4.8 (3) |
| N2i—N1—C10—N2 | 39.8 (2) | C1—C2—C3—C4 | 0.0 (4) |
| N2i—N1—C10—C1 | −146.05 (18) | Cl1—C2—C3—C4 | 178.2 (2) |
| N2—C10—C1—C2 | −121.8 (2) | C2—C3—C4—C5 | −0.9 (4) |
| N1—C10—C1—C2 | 63.9 (3) | C3—C4—C5—C6 | 0.2 (4) |
| N2—C10—C1—C6 | 59.8 (3) | C4—C5—C6—C1 | 1.2 (4) |
| N1—C10—C1—C6 | −114.4 (2) | C2—C1—C6—C5 | −2.0 (3) |
| C6—C1—C2—C3 | 1.3 (3) | C10—C1—C6—C5 | 176.5 (2) |
| C10—C1—C2—C3 | −177.05 (19) | | |
| Symmetry code: (i) −x, y, −z+1/2. |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1···N2ii | 0.88 (2) | 2.29 (2) | 3.052 (2) | 145 (2) |
| C6—H6···Cl1iii | 0.98 (2) | 2.83 (2) | 3.732 (2) | 154 (2) |
| Symmetry codes: (ii) x, −y+1, z−1/2; (iii) x, −y+1, z+1/2. |
(II) 3,5-bis(2-chlorophenyl)-4-amino-1
H-1,2,4-triazole
top
Crystal data top
| C14H10Cl2N4 | F(000) = 1248 |
| Mr = 305.16 | Dx = 1.458 Mg m−3 |
| Orthorhombic, Pbca | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2ac 2ab | Cell parameters from 29 reflections |
| a = 12.3382 (14) Å | θ = 4.1–10.3° |
| b = 8.6777 (9) Å | µ = 0.46 mm−1 |
| c = 25.968 (4) Å | T = 293 K |
| V = 2780.3 (6) Å3 | Prism, colourless |
| Z = 8 | 0.49 × 0.18 × 0.12 mm |
Data collection top
Siemens, P3
diffractometer | 1662 reflections with I > 2σ(I) |
| Radiation source: fine-focus sealed tube | Rint = 0.000 |
| Graphite monochromator | θmax = 25.1°, θmin = 2.3° |
| profile data from ω–2θ scans | h = 0→14 |
Absorption correction: gaussian
SHELX76 (Sheldrick, 1976) | k = 0→10 |
| Tmin = 0.913, Tmax = 0.952 | l = −30→0 |
| 2461 measured reflections | 2 standard reflections every 70 reflections |
| 2461 independent reflections | intensity decay: 0.0% |
Refinement top
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.036 | All H-atom parameters refined |
| wR(F2) = 0.098 | w = 1/[σ2(Fo2) + (0.0497P)2]
where P = (Fo2 + 2Fc2)/3 |
| S = 1.02 | (Δ/σ)max < 0.001 |
| 2461 reflections | Δρmax = 0.22 e Å−3 |
| 222 parameters | Δρmin = −0.23 e Å−3 |
| 0 restraints | Extinction correction: SHELXL97, Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| Primary atom site location: structure-invariant direct methods | Extinction coefficient: 0.0016 (4) |
Crystal data top
| C14H10Cl2N4 | V = 2780.3 (6) Å3 |
| Mr = 305.16 | Z = 8 |
| Orthorhombic, Pbca | Mo Kα radiation |
| a = 12.3382 (14) Å | µ = 0.46 mm−1 |
| b = 8.6777 (9) Å | T = 293 K |
| c = 25.968 (4) Å | 0.49 × 0.18 × 0.12 mm |
Data collection top
Siemens, P3
diffractometer | 1662 reflections with I > 2σ(I) |
Absorption correction: gaussian
SHELX76 (Sheldrick, 1976) | Rint = 0.000 |
| Tmin = 0.913, Tmax = 0.952 | 2 standard reflections every 70 reflections |
| 2461 measured reflections | intensity decay: 0.0% |
| 2461 independent reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.036 | 0 restraints |
| wR(F2) = 0.098 | All H-atom parameters refined |
| S = 1.02 | Δρmax = 0.22 e Å−3 |
| 2461 reflections | Δρmin = −0.23 e Å−3 |
| 222 parameters | |
Special details top
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| Cl1 | 0.50422 (5) | 0.13786 (7) | 0.26922 (2) | 0.0503 (2) | |
| Cl2 | 0.48752 (7) | 0.13503 (11) | 0.43241 (3) | 0.0908 (3) | |
| N1 | 0.78855 (15) | 0.2046 (2) | 0.38776 (7) | 0.0518 (6) | |
| N2 | 0.79746 (15) | 0.1982 (2) | 0.33424 (7) | 0.0473 (5) | |
| N3 | 0.65026 (13) | 0.3238 (2) | 0.35401 (6) | 0.0343 (4) | |
| N4 | 0.55912 (17) | 0.4213 (3) | 0.35109 (8) | 0.0416 (5) | |
| C1 | 0.71345 (16) | 0.2705 (2) | 0.31492 (8) | 0.0362 (5) | |
| C2 | 0.69952 (17) | 0.2803 (2) | 0.39847 (8) | 0.0396 (5) | |
| C11 | 0.69218 (16) | 0.2990 (2) | 0.26003 (8) | 0.0350 (5) | |
| C12 | 0.59812 (17) | 0.2490 (2) | 0.23554 (8) | 0.0367 (5) | |
| C13 | 0.5780 (2) | 0.2813 (3) | 0.18424 (9) | 0.0462 (6) | |
| C14 | 0.6538 (2) | 0.3637 (3) | 0.15663 (9) | 0.0515 (7) | |
| C15 | 0.7480 (2) | 0.4125 (3) | 0.17992 (9) | 0.0564 (7) | |
| C16 | 0.76670 (19) | 0.3809 (3) | 0.23107 (9) | 0.0454 (6) | |
| C21 | 0.65961 (19) | 0.3180 (3) | 0.45040 (8) | 0.0432 (6) | |
| C22 | 0.5635 (2) | 0.2598 (3) | 0.46946 (9) | 0.0535 (7) | |
| C23 | 0.5276 (3) | 0.2992 (4) | 0.51846 (12) | 0.0696 (9) | |
| C24 | 0.5879 (3) | 0.3959 (4) | 0.54759 (11) | 0.0777 (11) | |
| C25 | 0.6840 (3) | 0.4539 (4) | 0.53002 (11) | 0.0760 (9) | |
| C26 | 0.7199 (2) | 0.4152 (3) | 0.48162 (10) | 0.0587 (7) | |
| H4A | 0.512 (2) | 0.375 (3) | 0.3340 (10) | 0.065 (9)* | |
| H4B | 0.586 (2) | 0.508 (3) | 0.3383 (10) | 0.071 (10)* | |
| H13 | 0.5124 (18) | 0.251 (3) | 0.1703 (9) | 0.047 (7)* | |
| H14 | 0.6408 (19) | 0.383 (3) | 0.1202 (10) | 0.064 (8)* | |
| H15 | 0.800 (2) | 0.471 (3) | 0.1614 (10) | 0.070 (8)* | |
| H16 | 0.8318 (18) | 0.414 (2) | 0.2493 (8) | 0.045 (6)* | |
| H23 | 0.460 (2) | 0.262 (3) | 0.5265 (11) | 0.077 (10)* | |
| H24 | 0.561 (2) | 0.425 (3) | 0.5811 (12) | 0.086 (10)* | |
| H25 | 0.730 (2) | 0.529 (4) | 0.5505 (12) | 0.096 (11)* | |
| H26 | 0.789 (2) | 0.454 (3) | 0.4679 (10) | 0.064 (8)* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| Cl1 | 0.0436 (3) | 0.0512 (4) | 0.0561 (4) | −0.0137 (3) | 0.0032 (3) | −0.0011 (3) |
| Cl2 | 0.0859 (6) | 0.0964 (6) | 0.0902 (6) | −0.0431 (5) | 0.0163 (5) | −0.0045 (5) |
| N1 | 0.0455 (12) | 0.0693 (14) | 0.0407 (12) | 0.0152 (11) | −0.0017 (9) | 0.0066 (10) |
| N2 | 0.0387 (11) | 0.0658 (13) | 0.0375 (11) | 0.0138 (10) | 0.0020 (8) | 0.0034 (10) |
| N3 | 0.0303 (9) | 0.0368 (10) | 0.0356 (10) | 0.0053 (8) | −0.0002 (8) | 0.0008 (8) |
| N4 | 0.0341 (11) | 0.0426 (12) | 0.0482 (12) | 0.0070 (10) | −0.0028 (10) | −0.0031 (11) |
| C1 | 0.0311 (11) | 0.0391 (13) | 0.0384 (12) | 0.0000 (10) | 0.0030 (9) | 0.0001 (10) |
| C2 | 0.0370 (12) | 0.0451 (13) | 0.0367 (13) | 0.0033 (10) | −0.0009 (10) | 0.0035 (11) |
| C11 | 0.0335 (11) | 0.0344 (11) | 0.0371 (13) | 0.0044 (10) | 0.0010 (10) | −0.0034 (10) |
| C12 | 0.0377 (12) | 0.0291 (11) | 0.0435 (13) | 0.0004 (9) | 0.0029 (11) | −0.0021 (10) |
| C13 | 0.0477 (14) | 0.0474 (15) | 0.0433 (14) | 0.0014 (12) | −0.0081 (12) | −0.0040 (12) |
| C14 | 0.0642 (17) | 0.0547 (16) | 0.0357 (13) | 0.0010 (14) | −0.0017 (12) | 0.0034 (12) |
| C15 | 0.0608 (16) | 0.0624 (18) | 0.0461 (14) | −0.0128 (14) | 0.0098 (14) | 0.0090 (13) |
| C16 | 0.0376 (13) | 0.0527 (15) | 0.0458 (12) | −0.0081 (12) | 0.0040 (11) | 0.0002 (12) |
| C21 | 0.0476 (14) | 0.0454 (14) | 0.0364 (13) | 0.0096 (12) | −0.0002 (11) | 0.0049 (11) |
| C22 | 0.0579 (16) | 0.0561 (17) | 0.0464 (15) | 0.0051 (13) | 0.0071 (12) | 0.0101 (12) |
| C23 | 0.074 (2) | 0.084 (2) | 0.0504 (18) | 0.0169 (19) | 0.0204 (16) | 0.0230 (17) |
| C24 | 0.107 (3) | 0.090 (3) | 0.0355 (17) | 0.038 (2) | 0.0059 (18) | 0.0045 (17) |
| C25 | 0.096 (2) | 0.088 (2) | 0.0443 (18) | 0.012 (2) | −0.0170 (18) | −0.0105 (17) |
| C26 | 0.0573 (17) | 0.074 (2) | 0.0444 (15) | 0.0060 (16) | −0.0094 (13) | 0.0007 (14) |
Geometric parameters (Å, º) top
| Cl1—C12 | 1.743 (2) | C14—C15 | 1.376 (4) |
| Cl2—C22 | 1.725 (3) | C14—H14 | 0.97 (3) |
| N1—C2 | 1.310 (3) | C15—C16 | 1.376 (3) |
| N1—N2 | 1.395 (3) | C15—H15 | 0.95 (3) |
| N2—C1 | 1.311 (3) | C16—H16 | 0.98 (2) |
| N3—C2 | 1.358 (2) | C2—C21 | 1.473 (3) |
| N3—C1 | 1.361 (3) | C21—C22 | 1.381 (3) |
| N3—N4 | 1.410 (2) | C21—C26 | 1.387 (3) |
| N4—H4A | 0.83 (3) | C22—C23 | 1.390 (4) |
| N4—H4B | 0.88 (3) | C23—C24 | 1.352 (5) |
| C1—C11 | 1.470 (3) | C23—H23 | 0.92 (3) |
| C11—C16 | 1.384 (3) | C24—C25 | 1.367 (5) |
| C11—C12 | 1.393 (3) | C24—H24 | 0.96 (3) |
| C12—C13 | 1.384 (3) | C25—C26 | 1.374 (4) |
| C13—C14 | 1.379 (4) | C25—H25 | 1.02 (3) |
| C13—H13 | 0.93 (2) | C26—H26 | 0.98 (3) |
| | | |
| C2—N1—N2 | 107.30 (18) | C14—C15—H15 | 120.6 (16) |
| C1—N2—N1 | 107.46 (17) | C15—C16—C11 | 121.0 (2) |
| C2—N3—C1 | 106.45 (17) | C15—C16—H16 | 123.2 (13) |
| C2—N3—N4 | 124.73 (17) | C11—C16—H16 | 115.8 (13) |
| C1—N3—N4 | 128.39 (17) | N1—C2—N3 | 109.53 (18) |
| N3—N4—H4A | 107.3 (18) | N1—C2—C21 | 125.9 (2) |
| N3—N4—H4B | 103.6 (17) | N3—C2—C21 | 124.55 (19) |
| H4A—N4—H4B | 118 (2) | C22—C21—C26 | 118.3 (2) |
| N2—C1—N3 | 109.26 (19) | C22—C21—C2 | 122.3 (2) |
| N2—C1—C11 | 126.33 (19) | C26—C21—C2 | 119.4 (2) |
| N3—C1—C11 | 124.31 (18) | C21—C22—C23 | 120.8 (3) |
| C16—C11—C12 | 117.74 (19) | C21—C22—Cl2 | 119.75 (19) |
| C16—C11—C1 | 119.6 (2) | C23—C22—Cl2 | 119.5 (2) |
| C12—C11—C1 | 122.62 (19) | C24—C23—C22 | 119.3 (3) |
| C13—C12—C11 | 121.7 (2) | C24—C23—H23 | 126.2 (18) |
| C13—C12—Cl1 | 118.42 (18) | C22—C23—H23 | 114.2 (19) |
| C11—C12—Cl1 | 119.81 (16) | C23—C24—C25 | 121.3 (3) |
| C14—C13—C12 | 119.0 (2) | C23—C24—H24 | 118.9 (19) |
| C14—C13—H13 | 122.6 (15) | C25—C24—H24 | 119.9 (19) |
| C12—C13—H13 | 118.3 (15) | C24—C25—C26 | 119.7 (3) |
| C15—C14—C13 | 120.2 (2) | C24—C25—H25 | 123.2 (17) |
| C15—C14—H14 | 120.8 (15) | C26—C25—H25 | 117.0 (18) |
| C13—C14—H14 | 118.9 (15) | C25—C26—C21 | 120.7 (3) |
| C14—C15—C16 | 120.3 (2) | C25—C26—H26 | 121.7 (16) |
| C16—C15—H15 | 119.0 (16) | C21—C26—H26 | 117.6 (15) |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| N4—H4B···N2i | 0.88 (3) | 2.19 (3) | 3.016 (3) | 154 (2) |
| N4—H4B···N1i | 0.88 (3) | 2.64 (3) | 3.238 (3) | 126 (2) |
| N4—H4A···Cl1 | 0.83 (3) | 2.66 (3) | 3.321 (2) | 137 (2) |
| C16—H16···Cl1i | 0.98 (2) | 2.85 (2) | 3.734 (2) | 151 (2) |
| C24—H24···N4ii | 0.96 (3) | 2.66 (3) | 3.567 (4) | 157 (2) |
| C13—H13···N2iii | 0.93 (2) | 2.69 (2) | 3.568 (3) | 158 (2) |
| Symmetry codes: (i) −x+3/2, y+1/2, z; (ii) −x+1, −y+1, −z+1; (iii) x−1/2, y, −z+1/2. |
Experimental details
| | (I) | (II) |
| Crystal data |
| Chemical formula | C14H10Cl2N4 | C14H10Cl2N4 |
| Mr | 305.16 | 305.16 |
| Crystal system, space group | Monoclinic, C2/c | Orthorhombic, Pbca |
| Temperature (K) | 293 | 293 |
| a, b, c (Å) | 16.0020 (12), 9.4123 (8), 10.5752 (7) | 12.3382 (14), 8.6777 (9), 25.968 (4) |
| α, β, γ (°) | 90, 121.664 (5), 90 | 90, 90, 90 |
| V (Å3) | 1355.69 (18) | 2780.3 (6) |
| Z | 4 | 8 |
| Radiation type | Mo Kα | Mo Kα |
| µ (mm−1) | 0.47 | 0.46 |
| Crystal size (mm) | 0.62 × 0.09 × 0.09 | 0.49 × 0.18 × 0.12 |
| |
| Data collection |
| Diffractometer | Siemens, P3
diffractometer | Siemens, P3
diffractometer |
| Absorption correction | Gaussian
SHELX76 (Sheldrick, 1976) | Gaussian
SHELX76 (Sheldrick, 1976) |
| Tmin, Tmax | 0.936, 0.960 | 0.913, 0.952 |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 2496, 1203, 996 | 2461, 2461, 1662 |
| Rint | 0.014 | 0.000 |
| (sin θ/λ)max (Å−1) | 0.595 | 0.597 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.036, 0.099, 1.04 | 0.036, 0.098, 1.02 |
| No. of reflections | 1203 | 2461 |
| No. of parameters | 112 | 222 |
| H-atom treatment | All H-atom parameters refined | All H-atom parameters refined |
| Δρmax, Δρmin (e Å−3) | 0.39, −0.43 | 0.22, −0.23 |
Selected geometric parameters (Å, º) for (I) top| Cl1—C2 | 1.730 (2) | N2—C10 | 1.273 (2) |
| N1—C10 | 1.398 (2) | C10—C1 | 1.480 (3) |
| N1—N2i | 1.438 (2) | | |
| | | |
| C10—N1—N2i | 114.14 (15) | C2—C1—C10 | 123.78 (19) |
| C10—N2—N1i | 111.68 (16) | C6—C1—C10 | 118.17 (18) |
| N2—C10—N1 | 121.13 (18) | C1—C2—Cl1 | 120.20 (17) |
| N2—C10—C1 | 120.07 (17) | C3—C2—Cl1 | 119.56 (19) |
| N1—C10—C1 | 118.55 (16) | | |
| | | |
| N1i—N2—C10—N1 | 1.4 (3) | N2i—N1—C10—N2 | 39.8 (2) |
| N1i—N2—C10—C1 | −172.72 (17) | N2i—N1—C10—C1 | −146.05 (18) |
| Symmetry code: (i) −x, y, −z+1/2. |
Hydrogen-bond geometry (Å, º) for (I) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1···N2ii | 0.88 (2) | 2.29 (2) | 3.052 (2) | 145 (2) |
| C6—H6···Cl1iii | 0.98 (2) | 2.83 (2) | 3.732 (2) | 154 (2) |
| Symmetry codes: (ii) x, −y+1, z−1/2; (iii) x, −y+1, z+1/2. |
Selected geometric parameters (Å, º) for (II) top| Cl1—C12 | 1.743 (2) | N3—C2 | 1.358 (2) |
| Cl2—C22 | 1.725 (3) | N3—C1 | 1.361 (3) |
| N1—C2 | 1.310 (3) | N3—N4 | 1.410 (2) |
| N1—N2 | 1.395 (3) | C1—C11 | 1.470 (3) |
| N2—C1 | 1.311 (3) | C2—C21 | 1.473 (3) |
| | | |
| C2—N1—N2 | 107.30 (18) | N2—C1—C11 | 126.33 (19) |
| C1—N2—N1 | 107.46 (17) | N3—C1—C11 | 124.31 (18) |
| C2—N3—C1 | 106.45 (17) | N1—C2—N3 | 109.53 (18) |
| C2—N3—N4 | 124.73 (17) | N1—C2—C21 | 125.9 (2) |
| C1—N3—N4 | 128.39 (17) | N3—C2—C21 | 124.55 (19) |
| N2—C1—N3 | 109.26 (19) | | |
Hydrogen-bond geometry (Å, º) for (II) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| N4—H4B···N2i | 0.88 (3) | 2.19 (3) | 3.016 (3) | 154 (2) |
| N4—H4A···Cl1 | 0.83 (3) | 2.66 (3) | 3.321 (2) | 137 (2) |
| C16—H16···Cl1i | 0.98 (2) | 2.85 (2) | 3.734 (2) | 151 (2) |
| C24—H24···N4ii | 0.96 (3) | 2.66 (3) | 3.567 (4) | 157 (2) |
| C13—H13···N2iii | 0.93 (2) | 2.69 (2) | 3.568 (3) | 158 (2) |
| Symmetry codes: (i) −x+3/2, y+1/2, z; (ii) −x+1, −y+1, −z+1; (iii) x−1/2, y, −z+1/2. |


 journal menu
journal menu organic compounds
organic compounds











![[Figure 1]](http://journals.iucr.org/c/issues/2004/01/00/sk1670/sk1670fig1thm.gif)
![[Figure 2]](http://journals.iucr.org/c/issues/2004/01/00/sk1670/sk1670fig2thm.gif)
![[Figure 3]](http://journals.iucr.org/c/issues/2004/01/00/sk1670/sk1670fig3thm.gif)
![[Figure 4]](http://journals.iucr.org/c/issues/2004/01/00/sk1670/sk1670fig4thm.gif)




The compound of general formulae C2H2N4(C6H4Cl)2 crystallizes as 3,6-bis(2-chlorophenyl)-1,4-dihydro-1,2,4,5-tetrazine, (I), in the form of yellow–orange needles. It undergoes irreversible isomerization above 353 K in acidic water solution, forming the structural isomer 3,5-bis (2-chlorophenyl)-4-amino-1,2,4-triazol, (II).
X-ray structure determination revealed that isomer (I) crystallizes in the monoclinic space group C2/c, while isomer (II) crystallizes in the orthorhombic space group Pbca. The molecular structures of (I) and (II), with the atom-numbering schemes, are shown in Figs. 1 and 2, respectively.
Since both molecules retain potential strong proton-donor groups [–NH in (I) and –NH2 in (II)] and proton acceptors (N and Cl atoms) we undertook a detailed analysis of the hydrogen-bonded networks. Analysis of intra- and intermolecular contacts occurring in crystalline isomers indicated that, in both cases, the weaker –CH donor groups are also important in the formation of the hydrogen-bonded network (Taylor & Kennard, 1982; Desiraju & Steiner, 1999). To attempt a consistent characterization of the differences and similarities of the solid-state structures of the studied compounds, we have used the Etter (1990, 1991) graph-set descriptors.
The molecule of (I) consists of a six-membered central tetrazine ring and two chlorophenyl rings. As a twofold axis passes through the centre of the tetrazine ring, the molecule exhibits crystallographic C2 point-group symmetry, so the chlorophenyl rings are symmetry equivalent. The central ring is folded along the N1—N1i vector and exhibits a boat conformation [puckering parameters Q = 0.513 (2)°, θ = 90.0 (2)° and ϕ = 175.7 (2)°; Cremer & Pople, 1975]. An analysis of structures deposited in the Cambridge Structural Database (Version 5.24 of April 2003; Allen, 2002) showed that a boat conformation is observed for all structurally characterized tetrazine derivatives. The chlorophenyl rings are twisted to one another with a dihedral angle of 77.94 (7)°. The Cl atoms lie on the same side of the C10—N2—C10i—N2i plane [symmetry code: (i) −x, y, 1/2 − z]. Atoms N1 and N1i lie above this plane, and atoms H1 and H1i are located in equatorial positions. Such an arrangement enables the formation of a pair of strong intermolecular N—H···N hydrogen bonds between centrosymmetrically related neighbouring molecules [H···N = 2.29 (2) Å, N···N = 3.052 (2) Å and N—H···N = 145 (2)°; hydrogen-bond motif R22(6) (Bernstein et al., 1995); motif a in Fig.3], leading to the formation of an infinite C(4)[R22(6)] chain of rings along the c axis.
The molecule of (II) consists of a five-membered (heterocyclic) aromatic triazole ring and two chlorophenyl rings (Ph1 and Ph2). The main difference from the molecule of (I) is the asymmetry of the molecule of (II), which is a result of the postions of the H atoms of the –NH2 amine group. They lie on the same side of the quasi-mirror plane that passes through the amine N atom and the ring N—N bond, and is perpendicular to the ring plane. Excluding these H atoms, the molecule displays almost Cm point-group symmetry. The main reason for the asymmetry of the molecule is the strong N4—H4B···N2i intermolecular hydrogen bond [H···N = 2.19 (3) Å, N···N = 3.016 (3) Å and N—H···N 154 (2)°]. The observed infinite zigzag C(5) chain (motif a in Fig. 4) is formed by molecules related by a b-glide plane. As atoms H4A and H4B are both directed towards ring Ph1, the formation of an intramolecular hydrogen bond between the N4—H4A donor group and atom Cl1 is observed. The proton–acceptor distance is 2.66 (3) Å and the N4—H4A···Cl1 angle is 137 (2)°. The corresponding graph-set descriptor is S(7) (motif b in Fig. 4). This interaction is also evidenced by the significantly different thermal parameters of atoms Cl1 and Cl2, the Ueq values being 0.0503 (2) and 0.0908 (3), respectively. The dihedral angles between the triazole plane and the planes of the chlorophenyl rings Ph1 and Ph2 are 60.06 (7) and 65.21 (8)°, respectively. The larger value for ring Ph2 is a consequence of the repulsion between atoms Cl2 and N4 [Cl2···N4 = 3.378 (3) Å].
The observed one-dimensional assemblies formed by strong hydrogen bonding are enforced by weaker intermolecular interactions. C—H···Cl contacts are observed along the main chains, between the chlorophenyl rings of adjacent molecules. The H···Cl distances [2.83 (3) Å in (I) and 2.85 (2) Å in (II)] typify relatively strong C—H···Cl bonding (Taylor & Kennard, 1982). The assigned graph-set descriptors are C(5) for both compounds (motif b in Fig. 3 and motif c in Fig. 4). Motifs a, b and c constitute primary graph sets given as N1 = C(5) C(4)[R22(6)] [for (I)] and N1= C(5)S(7) C(5) [for (II)] (Etter, et al., 1990). The first-level graph sets are different because of the presence of symmetry-equivalent atoms in (I) and intramolecular hydrogen bonding in (II). Secondary graph sets for intermolecular bonding are N2(ab) = R22(11) [(I)] and N2(ac) = R22(12) [(II)], indicating the similarity of the main structural features.
Detailed analysis of the molecular packing shows that the main chains form close-packed structures. In the case of (II), the zigzag chains are connected together via C24—H24···N4v and C13—H13···N2vi weak hydrogen bonds, with motifs R22(16) and C(6), respectively. There is no evidence of weak hydrogen bonding between the chains of (I), but by analogy to the crystal structure of (II), the long C3—H3···N1iv contact [H···N = 2.98 (3) Å] can be regarded as an atractive interaction. The same graph-set descriptor, C(6), is applicable.