
Both title compounds are derivatives of salicylic acid. 5-Formylsalicylic acid (systematic name: 5-formyl-2-hydroxybenzoic acid), C
8H
6O
4, possesses three good hydrogen-bond donors and/or acceptors coplanar with their attached benzene ring and abides very well by Etter's hydrogen-bond rules. Intermolecular O-H

O and some weak C-H
O hydrogen bonds link the molecules into a planar sheet. Reaction of this acid and
o-phenylenediamine in refluxing ethanol produced in high yield the new zwitterionic compound 5-(benzimidazolium-2-yl)salicylate [systematic name: 5-(1
H-benzimidazol-3-ium-2-yl)-2-hydroxybenzoate], C
14H
10N
2O
3. Each imidazolium N-H group and its adjacent salicyl C-H group chelate one carboxylate O atom
via hydrogen bonds, forming seven-membered rings. As a result of steric hindrance, the planes of the molecules within these pairs of hydrogen-bonded molecules are inclined to one another by
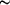
74°. There are also
![[pi]](/logos/entities/pi_rmgif.gif)
-
stacking interactions between the parallel planes of the imidazole ring and the benzene ring of the salicyl component of the adjacent molecule on one side and the benzimidazolium component of the molecule on the other side.
Supporting information
CCDC references: 810018; 810019
Compound (I) was prepared according to well established procedures (Duff &
Bills, 1932, 1934). Single crystals were obtained by vapour
diffusion of
diethyl ether into an ethanol solution. Analysis, calculated for C8H6O4:
C 57.84, H 3.64%; found: C 57.72, H 3.75%.
Compound (II) was synthesized as follows. A solution of o-phenyldiamine
(1.08 g, 0.01 mol) in ethanol (20 ml) was added to a solution of (I) (1.66 g,
0.01 mol) in ethanol (20 ml). The mixture was refluxed for 6 h. The flaxen
solid product was filtered, washed and recrystallized from ethanol (yield 2.14 g, 84%). Single crystals were obtained by vapour diffusion of diethyl ether
into an ethanol solution. Analysis, calculated for C14H10N2O3: C
66.14, H 3.96, N 11.02%; found: C 65.97, H 3.94, N 11.14%.
H atoms attached to C or N atoms were positioned geometrically and allowed to
ride on their parent atoms, with C—H = 0.93 Å and N—H = 0.86 Å, and
Uiso(H) = 1.2Ueq(C or N). H atoms bound to O atoms were
determined from difference Fourier maps and treated in the riding-model
approximation, with O—H = 0.82 Å and Uiso(H) =
1.5Ueq(O). [Please check added X—H distances]
For both compounds, data collection: APEX2 (Bruker, 2004); cell refinement: APEX2 (Bruker, 2004); data reduction: APEX2 (Bruker, 2004); program(s) used to solve structure: SHELXS97 (Sheldrick, 2008); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008); molecular graphics: ORTEP-3 (Farrugia, 1997); software used to prepare material for publication: PLATON (Spek, 2009).
(I) 5-Formyl-2-hydroxybenzoic acid
top
Crystal data top
| C8H6O4 | F(000) = 344 |
| Mr = 166.13 | Dx = 1.539 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2ybc | Cell parameters from 3041 reflections |
| a = 3.7762 (3) Å | θ = 3.0–26.0° |
| b = 16.3219 (11) Å | µ = 0.13 mm−1 |
| c = 11.6334 (8) Å | T = 298 K |
| β = 91.525 (5)° | Block, colourless |
| V = 716.77 (9) Å3 | 0.50 × 0.30 × 0.20 mm |
| Z = 4 | |
Data collection top
Bruker APEXII CCD area-detector
diffractometer | 1708 independent reflections |
| Radiation source: fine-focus sealed tube | 1202 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.043 |
| ϕ and ω scans | θmax = 28.0°, θmin = 2.2° |
Absorption correction: multi-scan
(SADABS; Bruker, 2002) | h = −4→3 |
| Tmin = 0.956, Tmax = 0.975 | k = −21→21 |
| 9607 measured reflections | l = −14→15 |
Refinement top
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.041 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.123 | H-atom parameters constrained |
| S = 1.03 | w = 1/[σ2(Fo2) + (0.0646P)2 + 0.0917P]
where P = (Fo2 + 2Fc2)/3 |
| 1699 reflections | (Δ/σ)max < 0.001 |
| 111 parameters | Δρmax = 0.22 e Å−3 |
| 0 restraints | Δρmin = −0.18 e Å−3 |
Crystal data top
| C8H6O4 | V = 716.77 (9) Å3 |
| Mr = 166.13 | Z = 4 |
| Monoclinic, P21/c | Mo Kα radiation |
| a = 3.7762 (3) Å | µ = 0.13 mm−1 |
| b = 16.3219 (11) Å | T = 298 K |
| c = 11.6334 (8) Å | 0.50 × 0.30 × 0.20 mm |
| β = 91.525 (5)° | |
Data collection top
Bruker APEXII CCD area-detector
diffractometer | 1708 independent reflections |
Absorption correction: multi-scan
(SADABS; Bruker, 2002) | 1202 reflections with I > 2σ(I) |
| Tmin = 0.956, Tmax = 0.975 | Rint = 0.043 |
| 9607 measured reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.041 | 0 restraints |
| wR(F2) = 0.123 | H-atom parameters constrained |
| S = 1.03 | Δρmax = 0.22 e Å−3 |
| 1699 reflections | Δρmin = −0.18 e Å−3 |
| 111 parameters | |
Special details top
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes)
are estimated using the full covariance matrix. The cell e.s.d.'s are taken
into account individually in the estimation of e.s.d.'s in distances, angles
and torsion angles; correlations between e.s.d.'s in cell parameters are only
used when they are defined by crystal symmetry. An approximate (isotropic)
treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s.
planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor
wR and goodness of fit S are based on F2, conventional
R-factors R are based on F, with F set to zero for
negative F2. The threshold expression of F2 >
σ(F2) is used only for calculating R-factors(gt) etc.
and is not relevant to the choice of reflections for refinement.
R-factors based on F2 are statistically about twice as large
as those based on F, and R-factors based on ALL data will be
even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| C1 | 0.7100 (4) | 0.69404 (8) | 0.09974 (11) | 0.0363 (3) | |
| C2 | 0.5386 (4) | 0.70240 (9) | −0.00863 (11) | 0.0396 (4) | |
| C3 | 0.4729 (4) | 0.78064 (9) | −0.05358 (12) | 0.0426 (4) | |
| H3A | 0.3625 | 0.7861 | −0.1256 | 0.051* | |
| C4 | 0.5691 (4) | 0.84907 (9) | 0.00710 (12) | 0.0414 (4) | |
| H4 | 0.5234 | 0.9007 | −0.0238 | 0.050* | |
| C5 | 0.7371 (4) | 0.84197 (8) | 0.11620 (11) | 0.0380 (3) | |
| C6 | 0.8046 (4) | 0.76454 (8) | 0.16036 (11) | 0.0370 (3) | |
| H6 | 0.9158 | 0.7596 | 0.2323 | 0.044* | |
| C7 | 0.7911 (4) | 0.61173 (9) | 0.14672 (12) | 0.0419 (4) | |
| C8 | 0.8511 (4) | 0.91348 (9) | 0.18176 (12) | 0.0442 (4) | |
| H8 | 0.9648 | 0.9049 | 0.2526 | 0.053* | |
| O1 | 0.4342 (3) | 0.63807 (7) | −0.07275 (9) | 0.0547 (3) | |
| H1 | 0.4742 | 0.5956 | −0.0371 | 0.082* | |
| O2 | 0.6998 (3) | 0.54837 (6) | 0.09905 (10) | 0.0589 (4) | |
| O3 | 0.9731 (3) | 0.61438 (6) | 0.24459 (9) | 0.0563 (4) | |
| H3 | 1.0152 | 0.5676 | 0.2667 | 0.085* | |
| O4 | 0.8078 (4) | 0.98366 (6) | 0.15020 (10) | 0.0641 (4) | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| C1 | 0.0407 (8) | 0.0318 (7) | 0.0363 (7) | −0.0006 (5) | −0.0050 (6) | 0.0005 (5) |
| C2 | 0.0442 (8) | 0.0383 (8) | 0.0359 (7) | −0.0032 (6) | −0.0051 (6) | −0.0056 (5) |
| C3 | 0.0491 (9) | 0.0445 (8) | 0.0336 (7) | −0.0003 (7) | −0.0123 (6) | 0.0013 (6) |
| C4 | 0.0481 (9) | 0.0355 (7) | 0.0401 (7) | 0.0019 (6) | −0.0085 (6) | 0.0057 (6) |
| C5 | 0.0427 (8) | 0.0324 (7) | 0.0386 (7) | −0.0006 (6) | −0.0064 (6) | −0.0006 (5) |
| C6 | 0.0430 (8) | 0.0352 (7) | 0.0324 (6) | 0.0001 (6) | −0.0085 (5) | 0.0012 (5) |
| C7 | 0.0517 (9) | 0.0338 (7) | 0.0397 (7) | −0.0015 (6) | −0.0054 (6) | −0.0006 (6) |
| C8 | 0.0538 (9) | 0.0369 (8) | 0.0413 (8) | −0.0007 (7) | −0.0120 (6) | −0.0002 (6) |
| O1 | 0.0784 (8) | 0.0405 (6) | 0.0441 (6) | −0.0048 (5) | −0.0198 (5) | −0.0069 (4) |
| O2 | 0.0845 (9) | 0.0331 (6) | 0.0581 (7) | −0.0037 (5) | −0.0174 (6) | −0.0054 (5) |
| O3 | 0.0860 (9) | 0.0317 (5) | 0.0499 (6) | 0.0037 (5) | −0.0255 (6) | 0.0030 (4) |
| O4 | 0.0975 (10) | 0.0308 (6) | 0.0622 (7) | −0.0027 (6) | −0.0305 (6) | −0.0020 (5) |
Geometric parameters (Å, º) top
| C1—C6 | 1.3913 (18) | C5—C6 | 1.3854 (18) |
| C1—C2 | 1.408 (2) | C5—C8 | 1.4532 (19) |
| C1—C7 | 1.4795 (19) | C6—H6 | 0.9300 |
| C2—O1 | 1.3411 (16) | C7—O2 | 1.2187 (16) |
| C2—C3 | 1.400 (2) | C7—O3 | 1.3149 (17) |
| C3—C4 | 1.365 (2) | C8—O4 | 1.2126 (17) |
| C3—H3A | 0.9300 | C8—H8 | 0.9300 |
| C4—C5 | 1.4086 (18) | O1—H1 | 0.8200 |
| C4—H4 | 0.9300 | O3—H3 | 0.8200 |
| | | |
| C6—C1—C2 | 118.64 (12) | C6—C5—C8 | 119.29 (12) |
| C6—C1—C7 | 121.06 (12) | C4—C5—C8 | 121.80 (12) |
| C2—C1—C7 | 120.29 (12) | C5—C6—C1 | 121.62 (12) |
| O1—C2—C3 | 117.39 (12) | C5—C6—H6 | 119.2 |
| O1—C2—C1 | 122.90 (13) | C1—C6—H6 | 119.2 |
| C3—C2—C1 | 119.71 (12) | O2—C7—O3 | 123.84 (14) |
| C4—C3—C2 | 120.74 (12) | O2—C7—C1 | 123.31 (14) |
| C4—C3—H3A | 119.6 | O3—C7—C1 | 112.85 (12) |
| C2—C3—H3A | 119.6 | O4—C8—C5 | 124.34 (13) |
| C3—C4—C5 | 120.39 (13) | O4—C8—H8 | 117.8 |
| C3—C4—H4 | 119.8 | C5—C8—H8 | 117.8 |
| C5—C4—H4 | 119.8 | C2—O1—H1 | 109.5 |
| C6—C5—C4 | 118.89 (12) | C7—O3—H3 | 109.5 |
| | | |
| C6—C1—C2—O1 | −179.28 (13) | C8—C5—C6—C1 | −178.20 (13) |
| C7—C1—C2—O1 | 1.6 (2) | C2—C1—C6—C5 | −0.6 (2) |
| C6—C1—C2—C3 | 1.1 (2) | C7—C1—C6—C5 | 178.56 (12) |
| C7—C1—C2—C3 | −178.11 (13) | C6—C1—C7—O2 | 176.71 (14) |
| O1—C2—C3—C4 | 179.49 (13) | C2—C1—C7—O2 | −4.1 (2) |
| C1—C2—C3—C4 | −0.8 (2) | C6—C1—C7—O3 | −3.7 (2) |
| C2—C3—C4—C5 | 0.1 (2) | C2—C1—C7—O3 | 175.47 (13) |
| C3—C4—C5—C6 | 0.3 (2) | C6—C5—C8—O4 | 179.28 (14) |
| C3—C4—C5—C8 | 178.39 (14) | C4—C5—C8—O4 | 1.2 (2) |
| C4—C5—C6—C1 | −0.1 (2) | | |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| O3—H3···O4i | 0.82 | 1.80 | 2.5848 (15) | 161 |
| O1—H1···O2ii | 0.82 | 2.54 | 3.0988 (16) | 127 |
| O1—H1···O2 | 0.82 | 1.94 | 2.6528 (15) | 145 |
| C3—H3A···O3iii | 0.93 | 2.63 | 3.4315 (18) | 144 |
| C4—H4···O4iv | 0.93 | 2.68 | 3.5641 (18) | 158 |
| C8—H8···O1v | 0.93 | 2.75 | 3.6576 (18) | 165 |
| Symmetry codes: (i) −x+2, y−1/2, −z+1/2; (ii) −x+1, −y+1, −z; (iii) x−1, −y+3/2, z−1/2; (iv) −x+1, −y+2, −z; (v) x+1, −y+3/2, z+1/2. |
(II) 5-(1
H-benzimidazol-3-ium-2-yl)-2-hydroxybenzoate
top
Crystal data top
| C14H10N2O3 | F(000) = 1056 |
| Mr = 254.24 | Dx = 1.366 Mg m−3 |
| Orthorhombic, Pbca | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2ac 2ab | Cell parameters from 5133 reflections |
| a = 16.2033 (4) Å | θ = 2.5–23.9° |
| b = 8.1005 (2) Å | µ = 0.10 mm−1 |
| c = 18.8425 (5) Å | T = 298 K |
| V = 2473.17 (11) Å3 | Block, yellow |
| Z = 8 | 0.35 × 0.30 × 0.20 mm |
Data collection top
Bruker APEXII CCD area-detector
diffractometer | 2949 independent reflections |
| Radiation source: fine-focus sealed tube | 2007 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.040 |
| ϕ and ω scans | θmax = 27.9°, θmin = 2.2° |
Absorption correction: multi-scan
(SADABS; Bruker, 2002) | h = −21→20 |
| Tmin = 0.966, Tmax = 0.981 | k = −10→10 |
| 23379 measured reflections | l = −24→24 |
Refinement top
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: inferred from neighbouring sites |
| R[F2 > 2σ(F2)] = 0.041 | H-atom parameters constrained |
| wR(F2) = 0.104 | w = 1/[σ2(Fo2) + (0.0442P)2 + 0.3316P]
where P = (Fo2 + 2Fc2)/3 |
| S = 1.05 | (Δ/σ)max < 0.001 |
| 2949 reflections | Δρmax = 0.20 e Å−3 |
| 174 parameters | Δρmin = −0.18 e Å−3 |
| 0 restraints | Extinction correction: SHELXL97 (Sheldrick, 2008), Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| Primary atom site location: structure-invariant direct methods | Extinction coefficient: 0.0029 (5) |
Crystal data top
| C14H10N2O3 | V = 2473.17 (11) Å3 |
| Mr = 254.24 | Z = 8 |
| Orthorhombic, Pbca | Mo Kα radiation |
| a = 16.2033 (4) Å | µ = 0.10 mm−1 |
| b = 8.1005 (2) Å | T = 298 K |
| c = 18.8425 (5) Å | 0.35 × 0.30 × 0.20 mm |
Data collection top
Bruker APEXII CCD area-detector
diffractometer | 2949 independent reflections |
Absorption correction: multi-scan
(SADABS; Bruker, 2002) | 2007 reflections with I > 2σ(I) |
| Tmin = 0.966, Tmax = 0.981 | Rint = 0.040 |
| 23379 measured reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.041 | 0 restraints |
| wR(F2) = 0.104 | H-atom parameters constrained |
| S = 1.05 | Δρmax = 0.20 e Å−3 |
| 2949 reflections | Δρmin = −0.18 e Å−3 |
| 174 parameters | |
Special details top
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes)
are estimated using the full covariance matrix. The cell e.s.d.'s are taken
into account individually in the estimation of e.s.d.'s in distances, angles
and torsion angles; correlations between e.s.d.'s in cell parameters are only
used when they are defined by crystal symmetry. An approximate (isotropic)
treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s.
planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor
wR and goodness of fit S are based on F2, conventional
R-factors R are based on F, with F set to zero for
negative F2. The threshold expression of F2 >
σ(F2) is used only for calculating R-factors(gt) etc.
and is not relevant to the choice of reflections for refinement.
R-factors based on F2 are statistically about twice as large
as those based on F, and R-factors based on ALL data will be
even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| C1 | 0.16371 (8) | 0.04754 (17) | 0.37266 (7) | 0.0406 (3) | |
| C2 | 0.09132 (9) | −0.0041 (2) | 0.40526 (9) | 0.0551 (4) | |
| H2 | 0.0397 | 0.0301 | 0.3895 | 0.066* | |
| C3 | 0.10025 (11) | −0.1088 (2) | 0.46222 (9) | 0.0662 (5) | |
| H3 | 0.0533 | −0.1473 | 0.4852 | 0.079* | |
| C4 | 0.17749 (11) | −0.1589 (2) | 0.48654 (9) | 0.0611 (5) | |
| H4 | 0.1806 | −0.2282 | 0.5258 | 0.073* | |
| C5 | 0.24948 (10) | −0.10858 (19) | 0.45404 (8) | 0.0496 (4) | |
| H5 | 0.3010 | −0.1419 | 0.4704 | 0.060* | |
| C6 | 0.24087 (8) | −0.00562 (17) | 0.39575 (7) | 0.0389 (3) | |
| C7 | 0.25852 (7) | 0.15783 (16) | 0.30215 (7) | 0.0349 (3) | |
| C8 | 0.29604 (7) | 0.25517 (16) | 0.24632 (7) | 0.0346 (3) | |
| C9 | 0.24716 (8) | 0.33425 (16) | 0.19499 (7) | 0.0395 (3) | |
| H9 | 0.1902 | 0.3208 | 0.1960 | 0.047* | |
| C10 | 0.28206 (8) | 0.43070 (18) | 0.14350 (7) | 0.0430 (3) | |
| H10 | 0.2487 | 0.4819 | 0.1099 | 0.052* | |
| C11 | 0.36763 (8) | 0.45280 (17) | 0.14105 (7) | 0.0417 (3) | |
| C12 | 0.41780 (7) | 0.37310 (16) | 0.19125 (7) | 0.0368 (3) | |
| C13 | 0.38124 (7) | 0.27549 (16) | 0.24272 (7) | 0.0366 (3) | |
| H13 | 0.4145 | 0.2221 | 0.2758 | 0.044* | |
| C14 | 0.50975 (8) | 0.39180 (18) | 0.19040 (8) | 0.0428 (3) | |
| N1 | 0.29802 (6) | 0.06433 (13) | 0.35000 (6) | 0.0384 (3) | |
| H1A | 0.3506 | 0.0500 | 0.3520 | 0.046* | |
| N2 | 0.17745 (6) | 0.14915 (13) | 0.31489 (6) | 0.0391 (3) | |
| H2A | 0.1398 | 0.1990 | 0.2909 | 0.047* | |
| O1 | 0.39888 (6) | 0.54939 (16) | 0.08991 (6) | 0.0643 (3) | |
| H1 | 0.4493 | 0.5517 | 0.0933 | 0.096* | |
| O2 | 0.55103 (5) | 0.31939 (14) | 0.23678 (6) | 0.0561 (3) | |
| O3 | 0.54029 (6) | 0.48116 (14) | 0.14132 (5) | 0.0560 (3) | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| C1 | 0.0358 (7) | 0.0418 (8) | 0.0442 (8) | −0.0015 (6) | 0.0056 (6) | −0.0070 (7) |
| C2 | 0.0384 (8) | 0.0653 (10) | 0.0617 (10) | −0.0027 (7) | 0.0125 (7) | −0.0009 (9) |
| C3 | 0.0618 (11) | 0.0726 (12) | 0.0642 (11) | −0.0120 (9) | 0.0239 (9) | 0.0002 (10) |
| C4 | 0.0767 (12) | 0.0578 (10) | 0.0488 (9) | −0.0037 (8) | 0.0126 (8) | 0.0031 (8) |
| C5 | 0.0577 (9) | 0.0487 (9) | 0.0424 (8) | 0.0045 (7) | −0.0006 (7) | −0.0057 (7) |
| C6 | 0.0375 (7) | 0.0389 (7) | 0.0402 (8) | −0.0009 (6) | 0.0036 (6) | −0.0096 (6) |
| C7 | 0.0261 (6) | 0.0366 (7) | 0.0421 (7) | 0.0009 (5) | 0.0004 (5) | −0.0084 (6) |
| C8 | 0.0268 (6) | 0.0349 (7) | 0.0422 (7) | 0.0012 (5) | 0.0005 (5) | −0.0067 (6) |
| C9 | 0.0243 (6) | 0.0463 (8) | 0.0480 (8) | −0.0005 (5) | −0.0020 (5) | −0.0063 (7) |
| C10 | 0.0309 (7) | 0.0522 (9) | 0.0458 (8) | 0.0027 (6) | −0.0059 (6) | 0.0006 (7) |
| C11 | 0.0344 (7) | 0.0480 (8) | 0.0427 (8) | −0.0020 (6) | 0.0014 (6) | −0.0008 (7) |
| C12 | 0.0257 (6) | 0.0422 (8) | 0.0423 (7) | 0.0001 (5) | 0.0007 (5) | −0.0058 (6) |
| C13 | 0.0274 (6) | 0.0396 (7) | 0.0428 (8) | 0.0039 (5) | −0.0013 (5) | −0.0047 (6) |
| C14 | 0.0275 (7) | 0.0525 (9) | 0.0483 (8) | −0.0018 (6) | 0.0031 (6) | −0.0089 (7) |
| N1 | 0.0274 (5) | 0.0433 (7) | 0.0444 (7) | 0.0026 (5) | 0.0001 (5) | −0.0054 (5) |
| N2 | 0.0254 (6) | 0.0452 (7) | 0.0468 (7) | 0.0015 (5) | 0.0012 (4) | −0.0025 (6) |
| O1 | 0.0412 (6) | 0.0887 (9) | 0.0629 (7) | −0.0101 (6) | −0.0014 (5) | 0.0266 (6) |
| O2 | 0.0262 (5) | 0.0793 (8) | 0.0627 (7) | 0.0067 (5) | −0.0011 (4) | 0.0058 (6) |
| O3 | 0.0314 (5) | 0.0771 (8) | 0.0596 (7) | −0.0121 (5) | 0.0033 (4) | 0.0062 (6) |
Geometric parameters (Å, º) top
| C1—N2 | 1.3827 (17) | C8—C9 | 1.4045 (18) |
| C1—C2 | 1.3888 (19) | C9—C10 | 1.3681 (19) |
| C1—C6 | 1.3920 (19) | C9—H9 | 0.9300 |
| C2—C3 | 1.375 (2) | C10—C11 | 1.3987 (18) |
| C2—H2 | 0.9300 | C10—H10 | 0.9300 |
| C3—C4 | 1.393 (2) | C11—O1 | 1.3405 (16) |
| C3—H3 | 0.9300 | C11—C12 | 1.4045 (19) |
| C4—C5 | 1.379 (2) | C12—C13 | 1.3844 (18) |
| C4—H4 | 0.9300 | C12—C14 | 1.4977 (17) |
| C5—C6 | 1.386 (2) | C13—H13 | 0.9300 |
| C5—H5 | 0.9300 | C14—O2 | 1.2469 (17) |
| C6—N1 | 1.3861 (17) | C14—O3 | 1.2745 (17) |
| C7—N2 | 1.3373 (15) | N1—H1A | 0.8600 |
| C7—N1 | 1.3402 (16) | N2—H2A | 0.8600 |
| C7—C8 | 1.4485 (18) | O1—H1 | 0.8200 |
| C8—C13 | 1.3919 (17) | | |
| | | |
| N2—C1—C2 | 131.57 (13) | C10—C9—H9 | 119.5 |
| N2—C1—C6 | 106.59 (11) | C8—C9—H9 | 119.5 |
| C2—C1—C6 | 121.82 (14) | C9—C10—C11 | 120.41 (13) |
| C3—C2—C1 | 116.24 (14) | C9—C10—H10 | 119.8 |
| C3—C2—H2 | 121.9 | C11—C10—H10 | 119.8 |
| C1—C2—H2 | 121.9 | O1—C11—C10 | 118.22 (12) |
| C2—C3—C4 | 122.05 (15) | O1—C11—C12 | 122.26 (12) |
| C2—C3—H3 | 119.0 | C10—C11—C12 | 119.52 (12) |
| C4—C3—H3 | 119.0 | C13—C12—C11 | 119.12 (12) |
| C5—C4—C3 | 121.86 (16) | C13—C12—C14 | 119.40 (12) |
| C5—C4—H4 | 119.1 | C11—C12—C14 | 121.48 (12) |
| C3—C4—H4 | 119.1 | C12—C13—C8 | 121.74 (12) |
| C4—C5—C6 | 116.39 (15) | C12—C13—H13 | 119.1 |
| C4—C5—H5 | 121.8 | C8—C13—H13 | 119.1 |
| C6—C5—H5 | 121.8 | O2—C14—O3 | 124.57 (12) |
| N1—C6—C5 | 132.17 (13) | O2—C14—C12 | 118.59 (13) |
| N1—C6—C1 | 106.22 (12) | O3—C14—C12 | 116.84 (13) |
| C5—C6—C1 | 121.61 (13) | C7—N1—C6 | 109.29 (11) |
| N2—C7—N1 | 108.58 (11) | C7—N1—H1A | 125.4 |
| N2—C7—C8 | 124.84 (12) | C6—N1—H1A | 125.4 |
| N1—C7—C8 | 126.58 (11) | C7—N2—C1 | 109.31 (11) |
| C13—C8—C9 | 118.15 (12) | C7—N2—H2A | 125.3 |
| C13—C8—C7 | 121.06 (12) | C1—N2—H2A | 125.3 |
| C9—C8—C7 | 120.78 (11) | C11—O1—H1 | 109.5 |
| C10—C9—C8 | 121.05 (12) | | |
| | | |
| N2—C1—C2—C3 | 179.26 (14) | O1—C11—C12—C13 | 179.83 (13) |
| C6—C1—C2—C3 | 0.9 (2) | C10—C11—C12—C13 | −0.7 (2) |
| C1—C2—C3—C4 | 0.7 (3) | O1—C11—C12—C14 | 0.0 (2) |
| C2—C3—C4—C5 | −1.2 (3) | C10—C11—C12—C14 | 179.47 (12) |
| C3—C4—C5—C6 | −0.1 (2) | C11—C12—C13—C8 | −0.44 (19) |
| C4—C5—C6—N1 | −178.22 (14) | C14—C12—C13—C8 | 179.39 (12) |
| C4—C5—C6—C1 | 1.7 (2) | C9—C8—C13—C12 | 1.28 (19) |
| N2—C1—C6—N1 | −0.96 (14) | C7—C8—C13—C12 | −177.57 (11) |
| C2—C1—C6—N1 | 177.74 (13) | C13—C12—C14—O2 | −0.56 (19) |
| N2—C1—C6—C5 | 179.07 (12) | C11—C12—C14—O2 | 179.27 (13) |
| C2—C1—C6—C5 | −2.2 (2) | C13—C12—C14—O3 | 179.05 (12) |
| N2—C7—C8—C13 | 171.03 (12) | C11—C12—C14—O3 | −1.11 (19) |
| N1—C7—C8—C13 | −7.9 (2) | N2—C7—N1—C6 | −0.74 (14) |
| N2—C7—C8—C9 | −7.78 (19) | C8—C7—N1—C6 | 178.31 (12) |
| N1—C7—C8—C9 | 173.32 (12) | C5—C6—N1—C7 | −178.98 (14) |
| C13—C8—C9—C10 | −0.99 (19) | C1—C6—N1—C7 | 1.06 (14) |
| C7—C8—C9—C10 | 177.86 (12) | N1—C7—N2—C1 | 0.11 (14) |
| C8—C9—C10—C11 | −0.1 (2) | C8—C7—N2—C1 | −178.96 (11) |
| C9—C10—C11—O1 | −179.52 (13) | C2—C1—N2—C7 | −177.98 (15) |
| C9—C10—C11—C12 | 1.0 (2) | C6—C1—N2—C7 | 0.54 (14) |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1A···O3i | 0.86 | 1.86 | 2.7102 (13) | 170 |
| N2—H2A···O2ii | 0.86 | 1.81 | 2.6544 (14) | 165 |
| O1—H1···O3 | 0.82 | 1.82 | 2.5482 (14) | 147 |
| C9—H9···O2ii | 0.93 | 2.58 | 3.4303 (16) | 151 |
| C13—H13···O3i | 0.93 | 2.60 | 3.4749 (17) | 156 |
| Symmetry codes: (i) −x+1, y−1/2, −z+1/2; (ii) x−1/2, y, −z+1/2. |
Experimental details
| | (I) | (II) |
| Crystal data |
| Chemical formula | C8H6O4 | C14H10N2O3 |
| Mr | 166.13 | 254.24 |
| Crystal system, space group | Monoclinic, P21/c | Orthorhombic, Pbca |
| Temperature (K) | 298 | 298 |
| a, b, c (Å) | 3.7762 (3), 16.3219 (11), 11.6334 (8) | 16.2033 (4), 8.1005 (2), 18.8425 (5) |
| α, β, γ (°) | 90, 91.525 (5), 90 | 90, 90, 90 |
| V (Å3) | 716.77 (9) | 2473.17 (11) |
| Z | 4 | 8 |
| Radiation type | Mo Kα | Mo Kα |
| µ (mm−1) | 0.13 | 0.10 |
| Crystal size (mm) | 0.50 × 0.30 × 0.20 | 0.35 × 0.30 × 0.20 |
| |
| Data collection |
| Diffractometer | Bruker APEXII CCD area-detector
diffractometer | Bruker APEXII CCD area-detector
diffractometer |
| Absorption correction | Multi-scan
(SADABS; Bruker, 2002) | Multi-scan
(SADABS; Bruker, 2002) |
| Tmin, Tmax | 0.956, 0.975 | 0.966, 0.981 |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 9607, 1708, 1202 | 23379, 2949, 2007 |
| Rint | 0.043 | 0.040 |
| (sin θ/λ)max (Å−1) | 0.660 | 0.658 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.041, 0.123, 1.03 | 0.041, 0.104, 1.05 |
| No. of reflections | 1699 | 2949 |
| No. of parameters | 111 | 174 |
| H-atom treatment | H-atom parameters constrained | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.22, −0.18 | 0.20, −0.18 |
Hydrogen-bond geometry (Å, º) for (I) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| O3—H3···O4i | 0.82 | 1.80 | 2.5848 (15) | 161 |
| O1—H1···O2ii | 0.82 | 2.54 | 3.0988 (16) | 127 |
| O1—H1···O2 | 0.82 | 1.94 | 2.6528 (15) | 145 |
| C3—H3A···O3iii | 0.93 | 2.63 | 3.4315 (18) | 144 |
| C4—H4···O4iv | 0.93 | 2.68 | 3.5641 (18) | 158 |
| C8—H8···O1v | 0.93 | 2.75 | 3.6576 (18) | 165 |
| Symmetry codes: (i) −x+2, y−1/2, −z+1/2; (ii) −x+1, −y+1, −z; (iii) x−1, −y+3/2, z−1/2; (iv) −x+1, −y+2, −z; (v) x+1, −y+3/2, z+1/2. |
Hydrogen-bond geometry (Å, º) for (II) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1A···O3i | 0.86 | 1.86 | 2.7102 (13) | 170 |
| N2—H2A···O2ii | 0.86 | 1.81 | 2.6544 (14) | 165 |
| O1—H1···O3 | 0.82 | 1.82 | 2.5482 (14) | 147 |
| C9—H9···O2ii | 0.93 | 2.58 | 3.4303 (16) | 151 |
| C13—H13···O3i | 0.93 | 2.60 | 3.4749 (17) | 156 |
| Symmetry codes: (i) −x+1, y−1/2, −z+1/2; (ii) x−1/2, y, −z+1/2. |


 journal menu
journal menu organic compounds
organic compounds










![[Figure 1]](http://journals.iucr.org/c/issues/2010/12/00/sk3389/sk3389fig1thm.gif)
![[Figure 2]](http://journals.iucr.org/c/issues/2010/12/00/sk3389/sk3389fig2thm.gif)
![[Figure 3]](http://journals.iucr.org/c/issues/2010/12/00/sk3389/sk3389fig3thm.gif)
![[Figure 4]](http://journals.iucr.org/c/issues/2010/12/00/sk3389/sk3389fig4thm.gif)




Salicylic acid is a plant hormone and widely used in organic synthesis (Hayat & Ahmad, 2007). This acid and many of its derivatives have medical applications, such as in anti-inflammatory treatments, easing aches and pains and reducing fevers. Formylation leads to separation of 3- and 5-formylsalicylic acids (Duff & Bills, 1932, 1934). The formyl group can then be used to react with various amines, which normally afford Schiff bases. The reaction of 5-formylsalicylic acid has received much less attention than that of 3-formylsalicylic acid. The crystal structure of 5-formylsalicylic acid, (I), has not been reported previously, although a cocrystal containing deprotonated (I), 2-aminopyridinium 5-formylsalicylate, has been published (Li et al., 2006).
A unique type of salicylic acid derivative, benzimidazolylsalicylic acids, was designed to combine both chemotherapeutic benzimidazole and salicylic acid moieties. These compounds have antimicrobial, cytotoxic and anthelmintic potential and were synthesized by interaction of 5,6-dimethyl- or 6-nitrobenzimidazoles with diazotized 5-aminosalicylic acid in the presence of cupric chloride (Dahiya & Pathak, 2007). The reported multi-step reaction is somewhat complicated. We found that when (I) is reacted with o-phenyldiamine in refluxing ethanol, the zwitterionic form of a benzimidazolylsalicylic acid, 5-(benzimidazolium-2-yl)salicylate, (II), could be obtained immediately in high yield. The imidazole ring in the new compound, (II), was formed through a one-step condensation between the formyl group of (I) and o-phenyldiamine, much more convenient than the method mentioned above (Dahiya & Pathak, 2007). Such an imidazole ring-enclosure approach involving aldehydes and aromatic orthodiamines has occasionally been utilized (Bindra & Elix, 1969; da Silva et al., 2010). However, in the case of 3-formylsalicylic acid, its reaction with o-phenyldiamine yielded a salen-type double Schiff base instead of an imidazolyl compound (Cheng & Liu, 2000; Lalehzari et al., 2008).
It is also notable that when (I) and o-phenyldiamine were mixed in refluxing syrupy phosphoric acid, which has a much higher boiling point than ethanol, it was the carboxyl not the aldehyde group of (I) that participated in the formation of imidazolyl ring closure, with the product being 2-(2-hydroxyphenyl)benzimidazole (Tong et al., 2005).
In (I), the parent phenyl ring and the three attached O-containing functional groups are almost exactly coplanar (Fig. 1). The interaction of the molecules can be analysed using the graph-set theory regarding the hydrogen-bond patterns of organic compounds, developed by Etter (1990). Some hydrogen-bond rules have been proposed, including one that all good H-atom donors and acceptors are used in hydrogen bonding, whereas less acidic H atoms may be used in hydrogen bonding when there are extra H-atom acceptors available after all the more acidic H atoms have found an acceptor. These rules apply very well in the case of (I). The hydroxyl, carboxyl and formyl groups are all good H-atom donors and/or acceptors. The hydroxyl and carboxyl groups, as H-atom donors, form both intra- and intermolecular O—H···O hydrogen bonds (Fig. 2, Table 1) and their hydrogen-bond patterns can be encoded as S(6) and R22(4), respectively (Etter, 1990). The resulting dimers are connected through intermolecular hydrogen bonding between the carboxyl donor and the formyl acceptor, leading to the formation of an infinite two-dimensional planar network parallel with the (102) plane. Some of the less acidic C—H groups also participate in intermolecular C—H···O hydrogen bonds, which are quite weak as the corresponding H···O distances are fairly long (Table 1), but which still agree with the description of C—H···O bonds (Steiner, 2003). Thus, a series of hydrogen-bond patterns is formed, including R33(9), R22(11), R32(7) and R22(10) (Fig. 2). Through these conventional and unconventional hydrogen bonds, one molecule of (I) is connected to six adjacent molecules in the same plane. These planar sheets are separated evenly by 3.406 Å, as there is a weak interaction between adjacent parallel phenyl rings (centroid-to-centroid distance 3.776 Å).
Compound (II) is the zwitterionic form of 5-(benzimidazol-2-yl)salicylic acid, in which the carboxyl group is deprotonated and an imidazole N atom is protonated, as shown in Fig. 3. The carboxylate and hydroxyl groups are coplanar with their attached phenyl ring, whereas the benzimidazolium moiety is slightly twisted from the salicyl moiety by 7.35°. Graph-set analysis is also applicable for the hydrogen bonding in (II). As shown in Fig. 4 and Table 2, the hydroxyl and carboxylate groups form an intramolecular S(6) hydrogen-bond pattern, similar to (I). Intermolecular N—H···O hydrogen bonds connect each molecule to four neighbouring molecules and form a two-dimensional network parallel to the (001) plane. Non-classical C—H···O hydrogen bonds also play an important role in the crystal packing. It is quite interesting that each of the two carboxylate O atoms is chelated by an N—H group and an adjacent salicyl C—H group, forming a seven-membered R21(7) ring. Due to steric requirements, these chelate cycles mean that each pair of hydrogen-bonded molecules is not coplanar but staggered, by an angle of ~74°. The three molecules on the same side of their connected molecule (Fig. 4) are aligned almost parallel and staggered. They are stabilized by two kinds of π–π stacking interactions between the imidazole ring and the benzene ring of either the salicyl or the benzoimidazolium moiety, as evident from the centroid-to-centroid distances: Cg(C1/C6/C7/N1/N2)···Cg(C8–C13)i = 3.5742 (8) Å, Cg(C1/C6/C7/N1/N2)···Cg(C1–C6)ii = 3.6978 (8) Å [symmetry codes: (i) -x + 1/2, y - 1/2, z; (ii) -x + 1/2, y + 1/2, z].