The title compound, C
30H
34O
8, crystallizes in the space group
P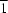
with one-half of a molecule in the asymmetric unit. A three-dimensional network is generated by OCH
3
![[pi]](/logos/entities/pi_rmgif.gif)
and CH
interactions. The conformation of the C-C bond exocyclic to the central benzene ring is different from that of every other known derivative. A comparison of the geometry of the title molecule and of its solid-state structure with other 2,4,6-trimethoxy-substituted PPV [
i.e. poly(
p-phenylenevinylene)] oligomers, in particular the isopropoxy-substituted compound, is provided.
Supporting information
CCDC reference: 273044
Compound (I) was prepared by the Wittig reaction of 2,4,6-trimethoxybenzaldehyde with 2,5-dimethoxy-p-xylylene-bis(triphenylphosphonium bromide), as described by Nowaczyk et al., 2005). Small crystal clusters were grown from hot ethanol solution, and a single fragment was used for the diffraction experiment
The low number of observed data and the low precision on the bond distances are a result of the crystal being small and weakly diffracting. The H atoms were placed at calculated positions and constrained with the SHELXL97 (Sheldrick, 1997) command AFIX 44 for aromatic and ethenylic H atoms, and AFIX 24 for methylene H atoms, which allow the H distances to refine, and with AFIX 138 for methoxy groups, which allows the methyl group to rotate and the H distances to refine, but which keeps the angles between the H atoms close to 109.5°. The filled volumes were calculated with PLATON (Spek, 2003).
Data collection: CAD-4 EXPRESS (Enraf–Nonius, 1994); cell refinement: CAD-4 EXPRESS; data reduction: XCAD4 (Harms, 1996); program(s) used to solve structure: SHELXS97 (Sheldrick, 1997); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics: ORTEP-3 (Farrugia, 1999) and Mercury (Bruno et al., 2002); software used to prepare material for publication: WinGX (Farrugia, 1997) and PLATON (Spek, 2003).
(
E,
E)-2,5-Dipropoxy-1,4-bis[2-(2,4,6-trimethoxyphenyl)ethenyl]benzene
top
Crystal data top
| C34H42O8 | F(000) = 310 |
| Mr = 578.68 | Dx = 1.292 Mg m−3 |
| Triclinic, P1 | Melting point: 168-169°C K |
| a = 6.842 (3) Å | Mo Kα radiation, λ = 0.71073 Å |
| b = 8.026 (2) Å | Cell parameters from 25 reflections |
| c = 14.215 (3) Å | θ = 4–12° |
| α = 73.99 (2)° | µ = 0.09 mm−1 |
| β = 82.82 (3)° | T = 293 K |
| γ = 86.17 (3)° | Block, yellow |
| V = 744.0 (4) Å3 | 0.1 × 0.1 × 0.1 mm |
| Z = 1 | |
Data collection top
Enraf–Nonius MACH3
diffractometer | Rint = 0.045 |
| Radiation source: fine-focus sealed tube | θmax = 25.0°, θmin = 1.5° |
| Graphite monochromator | h = 0→8 |
| ω/2θ scans | k = −9→9 |
| 2854 measured reflections | l = −16→16 |
| 2613 independent reflections | 3 standard reflections every 60 min |
| 1135 reflections with I > 2σ(I) | intensity decay: 6% |
Refinement top
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.087 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.272 | H-atom parameters constrained |
| S = 1.00 | w = 1/[σ2(Fo2) + (0.1376P)2]
where P = (Fo2 + 2Fc2)/3 |
| 2613 reflections | (Δ/σ)max < 0.001 |
| 212 parameters | Δρmax = 0.34 e Å−3 |
| 0 restraints | Δρmin = −0.33 e Å−3 |
Crystal data top
| C34H42O8 | γ = 86.17 (3)° |
| Mr = 578.68 | V = 744.0 (4) Å3 |
| Triclinic, P1 | Z = 1 |
| a = 6.842 (3) Å | Mo Kα radiation |
| b = 8.026 (2) Å | µ = 0.09 mm−1 |
| c = 14.215 (3) Å | T = 293 K |
| α = 73.99 (2)° | 0.1 × 0.1 × 0.1 mm |
| β = 82.82 (3)° | |
Data collection top
Enraf–Nonius MACH3
diffractometer | Rint = 0.045 |
| 2854 measured reflections | 3 standard reflections every 60 min |
| 2613 independent reflections | intensity decay: 6% |
| 1135 reflections with I > 2σ(I) | |
Refinement top
| R[F2 > 2σ(F2)] = 0.087 | 0 restraints |
| wR(F2) = 0.272 | H-atom parameters constrained |
| S = 1.00 | Δρmax = 0.34 e Å−3 |
| 2613 reflections | Δρmin = −0.33 e Å−3 |
| 212 parameters | |
Special details top
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| O2 | 0.4206 (6) | 0.1588 (5) | 0.0980 (3) | 0.0557 (12) | |
| O32 | 0.6856 (6) | −0.1854 (5) | 0.2853 (3) | 0.0586 (13) | |
| O34 | 1.2122 (6) | −0.2723 (5) | 0.4818 (3) | 0.0609 (13) | |
| O36 | 1.0559 (5) | 0.2918 (4) | 0.2720 (3) | 0.0528 (12) | |
| C1 | 0.6146 (7) | 0.4018 (7) | 0.0747 (4) | 0.0404 (14) | |
| C10 | 0.2691 (8) | −0.1121 (7) | 0.1294 (4) | 0.0466 (15) | |
| C11 | 0.1074 (9) | −0.2055 (8) | 0.1019 (5) | 0.063 (2) | |
| C2 | 0.4567 (8) | 0.3303 (6) | 0.0483 (4) | 0.0415 (14) | |
| C31 | 0.8687 (8) | 0.0536 (7) | 0.2775 (4) | 0.0398 (13) | |
| C32 | 0.8377 (8) | −0.1246 (7) | 0.3184 (4) | 0.0409 (14) | |
| C33 | 0.9511 (8) | −0.2301 (7) | 0.3860 (4) | 0.0482 (15) | |
| C34 | 1.1043 (8) | −0.1598 (7) | 0.4151 (4) | 0.0447 (15) | |
| C35 | 1.1455 (8) | 0.0146 (6) | 0.3778 (4) | 0.0419 (14) | |
| C36 | 1.0264 (8) | 0.1185 (6) | 0.3108 (4) | 0.0427 (14) | |
| C42 | 0.6556 (10) | −0.3659 (7) | 0.3139 (6) | 0.074 (2) | |
| C44 | 1.3719 (9) | −0.2018 (8) | 0.5137 (5) | 0.0625 (19) | |
| C46 | 1.2157 (10) | 0.3642 (8) | 0.3022 (5) | 0.068 (2) | |
| C6 | 0.6528 (8) | 0.5739 (7) | 0.0233 (4) | 0.0440 (15) | |
| C7 | 0.7473 (8) | 0.3137 (7) | 0.1479 (4) | 0.0449 (15) | |
| C8 | 0.7390 (8) | 0.1510 (7) | 0.2065 (4) | 0.0444 (15) | |
| C9 | 0.2664 (8) | 0.0754 (6) | 0.0699 (4) | 0.0436 (14) | |
| H10A | 0.2473 (10) | −0.1193 (7) | 0.201 (3) | 0.056* | |
| H10B | 0.399 (5) | −0.168 (2) | 0.1157 (6) | 0.056* | |
| H11A | −0.014 (4) | −0.153 (4) | 0.115 (3) | 0.095* | |
| H11B | 0.108 (4) | −0.320 (4) | 0.138 (3) | 0.095* | |
| H11C | 0.129 (4) | −0.200 (5) | 0.036 (2) | 0.095* | |
| H33 | 0.9257 (18) | −0.343 (7) | 0.4108 (16) | 0.058* | |
| H35 | 1.250 (6) | 0.061 (3) | 0.3970 (12) | 0.050* | |
| H42A | 0.771 (5) | −0.425 (2) | 0.292 (3) | 0.111* | |
| H42B | 0.546 (6) | −0.3893 (13) | 0.285 (3) | 0.111* | |
| H42C | 0.630 (7) | −0.405 (2) | 0.384 (3) | 0.111* | |
| H44A | 1.471 (4) | −0.147 (5) | 0.4532 (19) | 0.094* | |
| H44B | 1.443 (4) | −0.300 (3) | 0.563 (3) | 0.094* | |
| H44C | 1.3169 (19) | −0.107 (5) | 0.548 (3) | 0.094* | |
| H46A | 1.196 (3) | 0.344 (5) | 0.376 (3) | 0.102* | |
| H46B | 1.219 (4) | 0.493 (4) | 0.269 (3) | 0.102* | |
| H46C | 1.344 (4) | 0.306 (4) | 0.283 (3) | 0.102* | |
| H6 | 0.761 (6) | 0.627 (3) | 0.0390 (10) | 0.053* | |
| H7 | 0.846 (6) | 0.376 (4) | 0.1543 (6) | 0.054* | |
| H8 | 0.632 (6) | 0.090 (4) | 0.2008 (5) | 0.053* | |
| H9A | 0.2905 (10) | 0.0839 (7) | −0.006 (2) | 0.052* | |
| H9B | 0.129 (4) | 0.136 (2) | 0.0844 (6) | 0.052* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| C1 | 0.032 (3) | 0.040 (3) | 0.052 (3) | 0.004 (3) | −0.019 (3) | −0.014 (3) |
| C10 | 0.041 (3) | 0.044 (3) | 0.054 (4) | −0.002 (3) | −0.011 (3) | −0.008 (3) |
| C11 | 0.069 (5) | 0.053 (4) | 0.071 (5) | −0.017 (3) | −0.017 (4) | −0.015 (3) |
| C2 | 0.043 (4) | 0.028 (3) | 0.053 (4) | 0.000 (2) | −0.017 (3) | −0.007 (3) |
| C31 | 0.036 (3) | 0.040 (3) | 0.044 (3) | 0.001 (2) | −0.011 (3) | −0.010 (2) |
| C32 | 0.039 (3) | 0.041 (3) | 0.045 (3) | −0.008 (3) | −0.011 (3) | −0.011 (3) |
| C33 | 0.048 (4) | 0.038 (3) | 0.058 (4) | −0.004 (3) | −0.011 (3) | −0.009 (3) |
| C34 | 0.043 (3) | 0.046 (3) | 0.045 (3) | 0.005 (3) | −0.017 (3) | −0.008 (3) |
| C35 | 0.037 (3) | 0.040 (3) | 0.050 (3) | −0.006 (3) | −0.018 (3) | −0.007 (3) |
| C36 | 0.042 (3) | 0.031 (3) | 0.055 (4) | −0.003 (3) | −0.012 (3) | −0.008 (3) |
| C42 | 0.072 (5) | 0.038 (4) | 0.114 (6) | −0.018 (3) | −0.033 (4) | −0.010 (4) |
| C44 | 0.061 (4) | 0.059 (4) | 0.067 (4) | −0.007 (3) | −0.034 (4) | −0.002 (3) |
| C46 | 0.069 (5) | 0.048 (4) | 0.089 (5) | −0.019 (3) | −0.033 (4) | −0.005 (3) |
| C6 | 0.036 (3) | 0.039 (3) | 0.061 (4) | −0.004 (3) | −0.014 (3) | −0.015 (3) |
| C7 | 0.038 (3) | 0.041 (3) | 0.059 (4) | 0.001 (3) | −0.020 (3) | −0.013 (3) |
| C8 | 0.033 (3) | 0.044 (3) | 0.057 (4) | −0.001 (3) | −0.017 (3) | −0.009 (3) |
| C9 | 0.038 (3) | 0.040 (3) | 0.054 (4) | −0.006 (3) | −0.011 (3) | −0.009 (3) |
| O2 | 0.046 (2) | 0.045 (2) | 0.074 (3) | −0.0133 (18) | −0.030 (2) | 0.000 (2) |
| O32 | 0.052 (3) | 0.044 (2) | 0.082 (3) | −0.010 (2) | −0.030 (2) | −0.007 (2) |
| O34 | 0.065 (3) | 0.047 (2) | 0.068 (3) | −0.001 (2) | −0.038 (2) | 0.003 (2) |
| O36 | 0.048 (2) | 0.037 (2) | 0.074 (3) | −0.0062 (18) | −0.033 (2) | −0.0023 (19) |
Geometric parameters (Å, º) top
| O2—C2 | 1.386 (6) | C31—C8 | 1.454 (7) |
| O2—C9 | 1.438 (6) | C6—C2i | 1.367 (7) |
| O36—C36 | 1.366 (6) | C6—H6 | 0.9618 |
| O36—C46 | 1.435 (6) | C10—C9 | 1.509 (7) |
| C32—O32 | 1.364 (6) | C10—C11 | 1.523 (7) |
| C32—C33 | 1.375 (7) | C10—H10A | 0.9934 |
| C32—C31 | 1.407 (7) | C10—H10B | 0.9934 |
| O34—C34 | 1.369 (6) | C9—H9A | 1.0524 |
| O34—C44 | 1.437 (6) | C9—H9B | 1.0524 |
| O32—C42 | 1.414 (6) | C8—H8 | 0.9283 |
| C1—C2 | 1.394 (7) | C44—H44A | 1.0379 |
| C1—C6 | 1.397 (7) | C44—H44B | 1.0379 |
| C1—C7 | 1.467 (7) | C44—H44C | 1.0379 |
| C2—C6i | 1.367 (7) | C46—H46A | 1.0088 |
| C36—C35 | 1.390 (7) | C46—H46B | 1.0088 |
| C36—C31 | 1.414 (7) | C46—H46C | 1.0088 |
| C35—C34 | 1.388 (7) | C42—H42A | 0.9541 |
| C35—H35 | 0.9271 | C42—H42B | 0.9541 |
| C33—C34 | 1.380 (7) | C42—H42C | 0.9541 |
| C33—H33 | 0.8994 | C11—H11A | 0.9250 |
| C7—C8 | 1.342 (7) | C11—H11B | 0.9250 |
| C7—H7 | 0.8951 | C11—H11C | 0.9250 |
| | | |
| C2—O2—C9 | 118.4 (4) | O2—C9—C10 | 107.5 (4) |
| C36—O36—C46 | 117.8 (4) | O2—C9—H9A | 110.2 |
| O32—C32—C33 | 122.3 (4) | C10—C9—H9A | 110.2 |
| O32—C32—C31 | 114.5 (5) | O2—C9—H9B | 110.2 |
| C33—C32—C31 | 123.2 (5) | C10—C9—H9B | 110.2 |
| C34—O34—C44 | 117.1 (4) | H9A—C9—H9B | 108.5 |
| C32—O32—C42 | 118.8 (4) | C7—C8—C31 | 130.9 (5) |
| C2—C1—C6 | 115.9 (5) | C7—C8—H8 | 114.5 |
| C2—C1—C7 | 126.8 (5) | C31—C8—H8 | 114.5 |
| C6—C1—C7 | 117.2 (4) | O34—C44—H44A | 109.5 |
| C6i—C2—O2 | 123.1 (4) | O34—C44—H44B | 109.5 |
| C6i—C2—C1 | 120.8 (5) | H44A—C44—H44B | 109.5 |
| O2—C2—C1 | 116.0 (4) | O34—C44—H44C | 109.5 |
| O36—C36—C35 | 121.7 (4) | H44A—C44—H44C | 109.5 |
| O36—C36—C31 | 115.4 (4) | H44B—C44—H44C | 109.5 |
| C35—C36—C31 | 122.9 (5) | O36—C46—H46A | 109.5 |
| C34—C35—C36 | 118.3 (5) | O36—C46—H46B | 109.5 |
| C34—C35—H35 | 120.9 | H46A—C46—H46B | 109.5 |
| C36—C35—H35 | 120.9 | O36—C46—H46C | 109.5 |
| C32—C33—C34 | 119.0 (5) | H46A—C46—H46C | 109.5 |
| C32—C33—H33 | 120.5 | H46B—C46—H46C | 109.5 |
| C34—C33—H33 | 120.5 | O34—C34—C33 | 116.1 (5) |
| C8—C7—C1 | 128.0 (5) | O34—C34—C35 | 122.6 (5) |
| C8—C7—H7 | 116.0 | C33—C34—C35 | 121.3 (5) |
| C1—C7—H7 | 116.0 | O32—C42—H42A | 109.5 |
| C32—C31—C36 | 115.2 (5) | O32—C42—H42B | 109.5 |
| C32—C31—C8 | 117.9 (5) | H42A—C42—H42B | 109.5 |
| C36—C31—C8 | 126.9 (5) | O32—C42—H42C | 109.5 |
| C2i—C6—C1 | 123.2 (5) | H42A—C42—H42C | 109.5 |
| C2i—C6—H6 | 118.4 | H42B—C42—H42C | 109.5 |
| C1—C6—H6 | 118.4 | C10—C11—H11A | 109.5 |
| C9—C10—C11 | 109.2 (5) | C10—C11—H11B | 109.5 |
| C9—C10—H10A | 109.8 | H11A—C11—H11B | 109.5 |
| C11—C10—H10A | 109.8 | C10—C11—H11C | 109.5 |
| C9—C10—H10B | 109.8 | H11A—C11—H11C | 109.5 |
| C11—C10—H10B | 109.8 | H11B—C11—H11C | 109.5 |
| H10A—C10—H10B | 108.3 | | |
| | | |
| C33—C32—O32—C42 | −6.4 (9) | C33—C32—C31—C8 | 179.8 (5) |
| C31—C32—O32—C42 | 173.4 (6) | O36—C36—C31—C32 | −179.4 (5) |
| C9—O2—C2—C6i | 2.9 (8) | C35—C36—C31—C32 | 1.1 (8) |
| C9—O2—C2—C1 | −176.6 (5) | O36—C36—C31—C8 | 0.7 (9) |
| C6—C1—C2—C6i | −0.6 (9) | C35—C36—C31—C8 | −178.8 (5) |
| C7—C1—C2—C6i | −178.3 (5) | C2—C1—C6—C2i | 0.6 (9) |
| C6—C1—C2—O2 | 179.0 (5) | C7—C1—C6—C2i | 178.6 (5) |
| C7—C1—C2—O2 | 1.2 (9) | C2—O2—C9—C10 | 175.6 (5) |
| C46—O36—C36—C35 | 0.4 (8) | C11—C10—C9—O2 | 180.0 (5) |
| C46—O36—C36—C31 | −179.2 (5) | C1—C7—C8—C31 | 177.3 (6) |
| O36—C36—C35—C34 | 179.0 (5) | C32—C31—C8—C7 | −171.1 (6) |
| C31—C36—C35—C34 | −1.5 (9) | C36—C31—C8—C7 | 8.8 (10) |
| O32—C32—C33—C34 | 179.2 (6) | C44—O34—C34—C33 | −179.7 (6) |
| C31—C32—C33—C34 | −0.5 (9) | C44—O34—C34—C35 | 0.8 (8) |
| C2—C1—C7—C8 | −3.0 (10) | C32—C33—C34—O34 | −179.4 (5) |
| C6—C1—C7—C8 | 179.2 (6) | C32—C33—C34—C35 | 0.1 (9) |
| O32—C32—C31—C36 | −179.8 (5) | C36—C35—C34—O34 | −179.6 (5) |
| C33—C32—C31—C36 | 0.0 (8) | C36—C35—C34—C33 | 0.9 (9) |
| O32—C32—C31—C8 | 0.1 (8) | | |
| Symmetry code: (i) −x+1, −y+1, −z. |
Experimental details
| Crystal data |
| Chemical formula | C34H42O8 |
| Mr | 578.68 |
| Crystal system, space group | Triclinic, P1 |
| Temperature (K) | 293 |
| a, b, c (Å) | 6.842 (3), 8.026 (2), 14.215 (3) |
| α, β, γ (°) | 73.99 (2), 82.82 (3), 86.17 (3) |
| V (Å3) | 744.0 (4) |
| Z | 1 |
| Radiation type | Mo Kα |
| µ (mm−1) | 0.09 |
| Crystal size (mm) | 0.1 × 0.1 × 0.1 |
| |
| Data collection |
| Diffractometer | Enraf–Nonius MACH3
diffractometer |
| Absorption correction | – |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 2854, 2613, 1135 |
| Rint | 0.045 |
| (sin θ/λ)max (Å−1) | 0.595 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.087, 0.272, 1.00 |
| No. of reflections | 2613 |
| No. of parameters | 212 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.34, −0.33 |
Experimental (XRD) and calculated (DFT) C—H···n(O) distances, r(Å), between the H atoms on the olefinic link and the O atoms of the methoxy groups and angles, α(°), between the olefinic C—H bond and the O atom of the nearest methoxy group, of the [ap,ap] conformer of (1). top| Method | C7-H7···O36 | blank | C8-H8···O32 | blank | C8-H8···O2 | blank |
| blank | r | α | r | α | r | α |
| XRD | 2.169 | 121.17 | 2.198 | 102.55 | 2.030 | 126.30 |
| DFT | 2.166 | 118.78 | 2.224 | 104.41 | 2.260 | 121.88 |
| Original values of r and α were corrected by a normalization of the C—H bond lengths to 1.083 Å. |


 journal menu
journal menu organic compounds
organic compounds











![[Figure 1]](http://journals.iucr.org/c/issues/2005/05/00/sx1163/sx1163fig1thm.gif)
![[Figure 2]](http://journals.iucr.org/c/issues/2005/05/00/sx1163/sx1163fig2thm.gif)
![[Figure 3]](http://journals.iucr.org/c/issues/2005/05/00/sx1163/sx1163fig3thm.gif)
![[Figure 4]](http://journals.iucr.org/c/issues/2005/05/00/sx1163/sx1163fig4thm.gif)




In a recent study on the crystal packings of several 2,4,6-trimethoxy-substituted PPV oligomers with side chains of varying size on the central ring (Vande Velde et al., 2004), the claim was made that most of these compounds crystallize only with great difficulty since their solid-state structures are determined by weak OCH3···π interactions, which, owing to the discrete occurrence of methoxy groups and aromatic rings in the molecule, can only form networks spaced at specific intervals. For this to occur, the volume of the side chain on the central ring has to be well matched to the residual space between the molecules in order to generate long-range order. Four of these 2,4,6-trimethoxy-substituted compounds were crystallized and studied (Vande Velde et al., 2004), and four markedly different networks determining the packing in the solid were found. [The vast influence the particular substitution of the distyrylbenzene skeleton can have on the crystal structure was recently also demonstrated by Vande Velde et al. (2005)]. One feature the four previous compounds did have in common was the presence of stabilizing intramolecular interactions between the H atoms on the olefinic link and the O atoms in the methoxy groups, in a five-membered ring assembly, designated CH-n(O), as explained by Wu et al., 1996) and Vande Velde et al. (2004) (section 3.2 and Table 3). Theoretical calculations indicated that these interactions are stabilizing (Wu et al., 1996; Vande Velde et al., 2004). In the case of the title compound, (I), the situation is the same (see Table 1). For the isolated molecule, calculations at the DFT/B3LYP/6–31G* level (Frisch et al., 2001) indicate that the conformer with the `correct' conformation, [sp,sp], is 16.57 kJ.mol−1 more stable than the [ap,ap]-conformer (Fig. 1). Nevertheless, in the crystal, the latter less stable form is found. At the moment it is not clear what the precise reason for this is and further research is needed to shed more light on this matter. One possibility, in the context of the role of the volume of the side chain mentioned above, is that the reorientation of the central ring is a requirement for filling the space generated by the OCH3···π network in an efficient manner. For now, we limit ourselves mainly to the discussion of the crystal structure of (I), compare it with that of the isopropoxy isomer and only hint at a possible reason for the conformational anomaly.
The title compound (see Fig. 2 for structure and numbering scheme) crystallizes in space group P-1, with one-half of a molecule in the asymmetric unit and one molecule in the unit cell. Bond distances and angles contain no surprises; the aromatic bond distances display their usual slight variations as a result of steric hindrance around the highly substituted phenyl rings. The central and peripheral rings are at an angle of 6.3 (3)°, mainly owing to the torsion of the peripheral ring with respect to the double bond [ϕ = 6.8 (3)°]. The central ring and the double bond vector are in the same plane [ϕ = 0.5 (4)°].
The crystal structure can be described as flat interconnected sheets of molecules. For views of the structure and the resulting close contacts [C42···H44bi = 2.84 (3) Å and O32—C42···H44bi = 159.4 (8)°, H11b···H46bii = 2.23 (5) Å and C11—H11b···H46bii = 144 (3)°, and H42c···H44bi = 2.35 (4) Å and C42—H42c···H44bi = 112 (2)°; symmetry codes: (i) 2 − x, −1 − y, 1 − z; (ii) −1 + x, −1 + y, z] see Figs. 3 and 4.
The sheets themselves are connected to each other by OCH3-π interactions between the p-methoxy groups on the peripheral rings (A), and the peripheral rings (A) of the next molecule (Fig. 4). These contacts are given by C44—H44c···CgAiii = 2.86 (3) Å and 153.0 (18)° [symmetry code: (iii) 2 − x,-y,1 − z], in which CgA indicates the centroid of peripheral ring A.
After normalization of the positions of the terminal H atoms to 1.083 Å, the contact becomes C44—H44c···CgAiii (2.82 Å and 152.6°). This OCH3···π interaction is also expressed by the following close contacts (Fig. 3): C31···H44ciii = 2.77 (4) Å and C8—C31···H44ciii = 101.9 (7)°; C36···H44ciii = 2.88 (3) Å and O36—C36···H44ciii = 103.7 (9)°. There is one more CH···π contact that may contribute to the stabilization of the crystal structure, i.e. one between a β-H atom of a propoxy group and the peripheral ring (A) of the next molecule along the a axis (not shown in Figs. 3 and 4) [C10—H10a···CgAiv; 2.70 (3) Å and 145.1 (11)°; symmetry code: (iv) x − 1, y, z]. The latter distance becomes 2.63 Å after normalization of the position of the H atom.
The Cambridge Structural Database (Version 5.25 with November 2003 and April and July 2004 updates; Allen, 2002) lists only nine entries that have similar non-terminal β-H atoms of an alkyl group contacting the π-cloud of a methoxy-substituted benzene ring at a distance shorter than that in (I); in all cases but one the alkyl group is a substituent on a ring system. The fact that this type of contact is so rare leaves two possible interpretations. The first is that this particular contact is remarkably strong and contributes to the stabilization of the crystal structure. The second is that its occurrence is purely coincidental, and other interactions force it this close; in this case it is repulsive. No conclusive evidence for either of the two possibilities could be found here.
As mentioned before, the OCH3-π network is different from what was observed in the isopropoxy analogue (Vande Velde et al., 2004). In that structure, there are eight OCH3···π interactions per molecule, four between the para-methoxy groups of the peripheral rings (A) and the central rings (B), and four more between the ortho-methoxy groups on the peripheral ring (A) and the peripheral rings (A) [for a detailed description and figures we refer to Vande Velde et al. (2004)]. Additionally, all contact distances are shorter than the C44—H44···CgA contact observed in (I). The latter distance actually bears more similarity to that found for the compound with hexoxy substituents on the middle ring (Vande Velde et al., 2004), where a (weaker) contact exists between the ortho-methoxy groups of the peripheral rings (A) and the peripheral rings (A). For (I), the backbones of the molecules have apparently shifted even further over one another, bringing the para-methoxy group of the peripheral ring (A) into contact with the peripheral ring (A).
In the previous study (Vande Velde et al., 2004), we also compared two physical properties of the compounds in relation to the number and strength of the OCH3···π interactions present. Firstly, the melting point of (I) (441–442 K) fits nicely in the set of the four earlier compounds since it is nearly identical to that of the hexoxy compound (437–439 K). The number of OCH3···π interactions is indeed identical for both compounds without taking the CH···π contact of the propoxy group into account. Secondly, (I) has the highest density of all the 2,4,6-trimethoxy-substituted PPV oligomers we have measured so far (1.29 Mg m−3). Being an isomer of the isopropoxy compound, the comparison of the cell volumes and packing efficiency can be made directly; the cell volume of (I) is 744.0 (4) Å3 with a filled volume of 70.5%, while for the isopropoxy derivative these values are 776.8 (7) Å3 and 67.6%, respectively. This remarkably high packing efficiency may be a result of the flipping over of the central ring (B) with respect to the equilibrium (gas-phase) conformation. This results in the presence of the less stable [ap,ap]-conformer in the crystal; apparently, the energy required to force the molecules into the energetically less favourable conformer is provided by the van der Waals contact stabilization. It is likely that the solid-state structure of the [sp,sp]-conformer displays a large number of destabilizing interactions as a result of steric hindrance between the side chains of neighbouring molecules. As mentioned above, further research is necessary in order to investigate these issues to a greater extent. Unfortunately, the presence of the [ap,ap]-conformer makes treating the density as a function of the number of OCH3···π contacts impossible.
Finally, a TLS analysis (Shomaker et al., 1968) with the PLATON software (Spek, 2003) shows that the largest librational movement is along the long axis of the molecule, as is customary for this type of compound. The difference between the different directions is quite small for (I) (L1 = 4.28°2) and lies along an axis making an angle of 4.07° with the long axis of the molecule. L2 and L3 are 1.18 and 0.63°2, respectively.
In conclusion, (I) crystallizes in small clustered crystals, which display a hitherto unobserved packing based on OCH3···π interactions and CH···π interactions. In effect, the compound is a spectacular demonstration of how the moving of one methyl group in a large molecule can make a remarkable difference in the crystal packing of compounds in which good crystal synthons are absent and the main interactions available for forming the crystal structure are weak.