

 journal menu
journal menu





issue contents
September 2001 issue
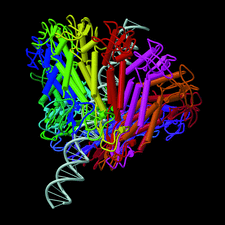
Cover illustration: The structure of the head-tail connector of bacteriophage ![[varphi]](/logos/entities/phiv_rmgif.gif) 29, including a model of DNA passing through its center. Each monomer is represented by a different color (p. 1260).
29, including a model of DNA passing through its center. Each monomer is represented by a different color (p. 1260).
research papers






The 2.1 Å structure of the isocitrate lyase from E. coli reveals a different domain structure to that seen in the enzyme from the eukaryote A. nidulans. However, the active sites of both enzymes remain closely related, including conformational changes affecting equivalent regions.
PDB reference: isocitrate lyase, 1igw

The structure solution of the apocrustacyanin A1 subunit of the lobster astaxanthin protein crustacyanin is described utilizing 2 Å X-rays to optimize the  signal to 11 electrons for xenon LI edge `SIROAS' phasing, together with enhanced sulfur anomalous scattering to determine the hand of the molecule. The structure has been refined at 1.4 Å resolution and is that of a typical lipocalin.
signal to 11 electrons for xenon LI edge `SIROAS' phasing, together with enhanced sulfur anomalous scattering to determine the hand of the molecule. The structure has been refined at 1.4 Å resolution and is that of a typical lipocalin.
PDB reference: lobster apocrustacyanin A1, 1h91
The structure of the C1 subunit of the carotenoid-binding protein α-crustacyanin has been determined using the anomalous scattering at 1.77 Å wavelength of the S atoms intrinsic to the native protein combined with a new ab initio phasing procedure. Although exploitation of the anomalous signal from S atoms has previously been demonstrated to be a successful phasing tool for a number of model proteins, this is to our knowledge the first time that this has been shown to be successful for such a large (40 kDa) previously unknown protein structure.
PDB reference: C1 subunit of α-crustacyanin, 1i4u

The X-ray crystal structure at 2.0 Å resolution of a DNA molecule complexed with the N-terminal fragment of Moloney murine leukemia virus reverse transcriptase (MMLV RT) has been determined. This method allows the study of nucleic acids in a unique and largely unfettered environment without the complicated lattice interactions typically observed in DNA-only crystal structures.
PDB reference: pseudo-16-mer DNA, 1ibj

The native and selenomethionine-substituted orotidine 5′-monophosphate decarboxylases crystallize with different symmetries. This is caused by different crystal contacts near the Met and SeMet residues.
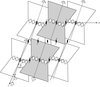
The head–tail connector of bacteriophage φ29, whose structure determination is described here, is the central component of a rotary motor that packages the genomic dsDNA into preformed proheads. Initial, 20 Å resolution, phases were obtained from a crude cryo-electron microscopic model, followed by non-crystallographic symmetry averaging, determination of heavy-atom sites and further averaging plus density modification.
The crystal structure of staphylococcal enterotoxin C2 has been determined at various pH levels (5.0–8.0) to study the effect of pH on the bound zinc.
The application of a conventional direct-method program, MULTAN88, using improved figures of merit, was able to generate and identify phase sets capable of development to a complete structure. The use of the density-modification procedure PERP led to a density map that could be routinely interpreted.
Single amino-acid substitutions in the contact regions of reaction centers from R. sphaeroides were found to lead to a significant decrease in the order of the protein crystals, as evident from both the size and the X-ray diffraction of the crystals.
crystallization papers

A complex of human CDK6 and a virus-encoded cyclin was crystallized in space group P6122 or P6522. The addition of small charged organic molecules proved to be essential for crystallization. The crystals diffract to at least 3.1 Å resolution.

Single-stranded DNA-binding (SSB) proteins are involved whenever the DNA duplex separates and thus play a key role in replication, transcription, recombination and repair. SSB from S. solfataricus differs in sequence from other SSB proteins characterized and has some unique biochemical properties. The expression, purification, crystallization and high-resolution data collection of this protein are reported.

Crystallization of a disulfide-bond reductant protein from B. japonicum and production of SeMet protein and crystals without using a methionine auxotroph or methionine-pathway inhibition are reported.
The dimer formed by the isolated N-terminal domain of riboflavin synthase is assumed to closely resemble the riboflavin-binding motif of the native enzyme. The crystallographic analysis of the domain in the presence of bound riboflavin provides an opportunity to acquire structural information on the riboflavin-binding site.

B. stearothermophilus L1 lipase was crystallized in two different crystal forms. Diffraction data for a native crystal and a derivative crystal were collected from strongly diffracting monoclinic crystals.
The truncated 1,3-1,4-β-glucanase (1,3-1,4-β-D-glucan 4-glucanohydrolase; E.C. 3.2.1.73) from F. succinogenes was crystallized in four different forms by the vapour-diffusion method.

A novel Fab fragment that prevents the binding of nerve-growth factor to its tyrosine kinase A receptor has been purified and crystallized; 1.8 Å resolution data have been collected using a synchrotron-radiation source. A clear molecular-replacement solution was obtained and the structure refinement is in progress.

The B. pertussis wlbD gene product is a putative UDP-GlcNAc 2′-epimerase. However, the biochemical transformation catalysed by this protein has not been confirmed. All essential active-site residues in the homologous E. coli rffE protein are present in the wlbD sequence, but key residues outside the active site involved in orientating an active-site helix are absent. A structure of the wlbD protein will help clarify the role of remote sodium binding in UDP-GlcNAc 2′-epimerase action.

The depressant insect toxin dITAP3 from the venom of the scorpion B. martensii Karsch has been crystallized (R3; a = b = 73.29, c = 68.90 Å). Diffraction data have been collected to 2.6 Å resolution using synchrotron X-rays.
An unusual trypsin inhibitor with two non-covalently bound chains, isolated from seeds of C. langsdorffii, was purified to homogeneity and crystallized. The crystals belong to the space group P41(3)212 and diffract to 1.8 Å resolution.

NAD kinase from M. tuberculosis H37Rv utilizes ATP or inorganic polyphosphate [poly(P)] as a phosphoryl donor for the phosphorylation of NAD. The NAD kinase overexpressed in E. coli was crystallized. This is the first report on the crystallographic data of an NAD kinase.

The catalytic subunit of S. cerevisiae acetohydroxyacid synthase (E.C. 4.1.3.18) has been crystallized and subjected to X-ray diffraction analysis using synchrotron radiation. Data to 2.7 Å resolution have been collected and preliminary analysis indicates that there is one dimer located in each asymmetric unit.

Crystals of LuxS a member of a family of proteins with a potential role in quorum sensing were obtained by the hanging-drop vapour-diffusion method, using ammonium sulfate as the precipitant. The crystals diffract to high resolution and belong to one of the enantiomorphic space groups P6122 or P6522, with approximate unit-cell parameters a = b = 63.6, c = 151.5 Å and one subunit in the asymmetric unit.

The crystallization and preliminary crystallographic study of phycocyanin from the thermophilic cyanobacterium S. elongatus is reported.

The crystallization and preliminary X-ray analysis of haemoglobin IV from the armored catfish L. anisitsi is reported.
A novel 40 kDa protein has been purified from dry secretions of the mammary gland of goats. The first 15 N-terminal residues were sequenced and showed a sequence identity of 30% to a novel 39 kDa whey protein from bovine mammary secretions. The protein was crystallized by the microdialysis method.
short communications
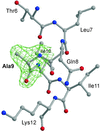
The crystal structure of a binding-deficient interleukin 4 mutant Glu9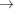 Ala has been solved at 2.1 Å resolution. Together with functional analyses this suggests a polar steering upon complex formation.
Ala has been solved at 2.1 Å resolution. Together with functional analyses this suggests a polar steering upon complex formation.
PDB reference: interleukin 4 mutant E9A, 1hzi
Threonine synthase crystals containing oxidized and reduced selenomethionine were used for multiple-wavelength anomalous dispersion; the comparison showed that uniformly oxidized selenomethionine strongly enhances the phasing power.
PDB reference: threonine synthase, 1e5x
A flexible web-accessible database for protein crystallography project management (LISA) has been developed using the open-source software MySQL and PHP4.
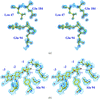
The 1.9 Å resolution structure of a mutant, E94A, of the Xyn11 from B. agaradhaerens in complex with xylotriose reveals the unusual 2,5B conformation at the catalytic centre first reported for the covalent xylobiosyl-enzyme intermediate, whose structure has also been extended to atomic (1.1 Å) resolution.

The use is presented of a histochemical procedure, the Karnovsky method, for easy and rapid identification of acetylcholinesterase crystals.

When applicable in crystallographic applications, the aggreement between two paired sets of structure factors is best assessed in terms of a correlation coefficient based on their full complex values, rather than the structure factor magnitudes alone as is commonly practised.
book reviews
Free 






