

 journal menu
journal menu




issue contents
December 2013 issue
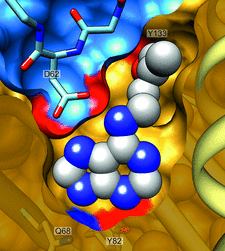
Cover illustration: The phytohormone cytokinin (spheres) is bound in the cavity of a plant PR-10 protein upon its dimerization (gold and blue), in a process that starts plant-microbe symbiosis (p. 2365).
scientific comment




Open  access
access
access
Deposition of crystallographic structures should be concurrent with or prior to manuscript submission for peer review, enabling validation and increasing reliability of the PDB.
letters to the editor
Open access
access
Open access
access
A response to the article by Jaskolski [(2013), Acta Cryst. D69, 1865–1866].
research papers

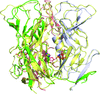
Horizontal gene transfer in some Staphyloccal phages is regulated by phage dUTPases, which are also responsible for maintaining genome integrity. Insights into the molecular mechanism of this regulation as well as into the enzymatic action of Φ11 phage dUTPase is provided by three-dimensional structural and enzyme kinetics studies.
PDB reference: φ11 phage dUTPase, 4gv8
High-resolution crystal structures of the designed calcium sensor CatchER revealed snapshots of calcium and gadolinium ions binding within the designed site in agreement with its fast kinetics.

The crystal structure of TenA from methicillin-resistant S. aureus, which is a thiaminase type II in the thiamine metabolism of the pathogen and may act as a transcriptional activator controlling the secretion of extracellular proteases, is reported. Mutagenic studies reveal the stabilizing effect of the tetrameric assembly against proteolytic digestion.
PDB reference: thiaminase II, 4fn6

The structure of L-ribulose 3-epimerase was determined and the unique structural features of the enzyme were compared with those of other ketose 3-epimerases.
PDB reference: L-ribulose 3-epimerase, 3vyl
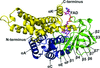
The crystal structure of AnaB, the prolyl-acyl carrier protein oxidase involved in anatoxin-a biosynthesis, is reported. The structural basis for the oxidase activity of this flavoprotein with an acyl-CoA dehydrogenase fold is described.
PDB reference: AnaB, 4irn
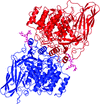
The crystal structure of a complex of two mulberry silkworm storage proteins, SP2 and SP3, has been determined. The presence of two different proteins in the native complex was detected crystallographically according to electron-density maps.
PDB reference: SP2–SP3 complex, 4l37
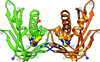
The crystal structures of complexes of M. truncatula nodulin 13 with four cytokinins, trans-zeatin, N6-isopentenyladenine, kinetin and N6-benzyladenine, show an unusual mode of dimerization of this PR-10-fold plant protein. The cytokinin-binding mode in the internal cavity of the protein is the same in each complex and resembles the pattern found in the cytokinin receptor protein.
Open access
access
Electron paramagnetic resonance (EPR) and online UV–visible absorption microspectrophotometry with X-ray crystallography have been used in a complementary manner to follow X-ray-induced disulfide-bond cleavage, to confirm a multi-track radiation-damage process and to develop a model of that process.
Open access
access
LigSearch is a web server for identifying ligands likely to bind to a given protein. It can be accessed at http://www.ebi.ac.uk/thornton-srv/databases/LigSearch.

The structure of the thrombin–HD22-27mer complex, solved at 2.4 Å resolution, reveals an intriguing duplex–quadruplex folding of the aptamer, which extensively interacts with the exosite II of the protein.
PDB reference: thrombin–HD22-27mer, 4i7y
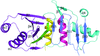
The Iml3 protein adopts a novel structure to dimerize with the Chl4 in kinetochore assembly.
PDB reference: Iml3, 4kr1

The structural and functional characterization of the N-terminal domain of the Toll-like receptor signalling adaptor TRIF/TICAM-1 is presented. The 2.22 Å resolution crystal structure was determined by selenomethionine-based SAD phasing using a protein containing two additional introduced methionines.
Open access
access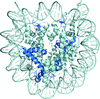
The crystal structures of human nucleosomes containing H2A.Z.1 and H2A.Z.2 have been determined. Structural polymorphisms were found in the L1 loop regions of H2A.Z.1 and H2A.Z.2 in the nucleosomes that are likely to be caused by their flexible nature.
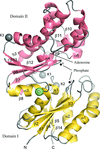
The crystal structure of a putative sugar kinase, MK0840, from M. kandleri has been solved in apo and nucleotide-bound forms at high resolution. Large domain movements are observed depending on the crystal form and the nucleotide state.

The design of a new protein fold by fusion of a heterodimer is described.
PDB reference: FliS-FliC fusion, 4iwb

Crystal structures of M. tuberculosis HisB and its substrate-bound and inhibitor-bound forms were determined.
Open access
access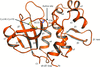
Crystal structures of type VI secretion system-associated immunity proteins, a peptidoglycan endopeptidase and a complex of the endopeptidase and its cognate immunity protein are reported together with assays of endopeptidase activity and functional assessment.
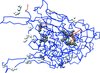
The crystal structure of a copper amine oxidase from A. globiformis was determined at 1.08 Å resolution. Diatomic molecules, most likely to be molecular oxygen, were detected along a route from the central cavity of the dimer interface to the active site.
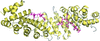
Nucleocytoplasmic trafficking of dUTPase, an enzyme essential for DNA integrity, is regulated by phosphorylation. The mechanism underlying this control is revealed by three-dimensional structures and cellular investigations.
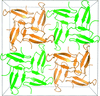
Two structures of a small protein with a defined tertiary fold, the isolated Pin1 WW domain, have been determined via racemic crystallization with small-molecule additives. These additives, which are either racemic or achiral, appear to stabilize a dynamic loop region of the structure.
Open access
access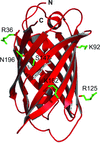
A strategy using a new split green fluorescent protein (GFP) as a modular binding partner to form stable protein complexes with a target protein is presented. The modular split GFP may open the way to rapidly creating crystallization variants.
Open access
access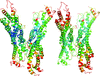
The anisotropy of crystals of glypican-1 was significantly reduced by controlled dehydration using the HC1 device, allowing the building of previously disordered parts of the structure.
PDB reference: glypican-1, 4bwe
Open access
access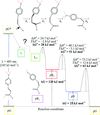
Barriers of activation within the photocycle of a photoactive protein were extracted from comprehensive time courses of time resolved crystallographic data collected at multiple temperature settings.
Open access
access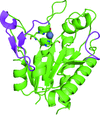
The structure of Rv3717 determined to 1.7 Å resolution by Pt-SAD phasing reveals a unique autolysin that lacks a cell-wall-binding domain. Rv3717 utilizes its net positive charge for substrate binding and exhibits activity towards a broad spectrum of substrate cell walls. Structural analysis reveals that Rv3717 utilizes a β-hairpin turn at its N-terminus to autoregulate its enzymatic activity.
PDB reference: Rv3717, 4lq6
Open access
access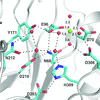
Human AP endonuclease 1 (APE1) belongs to the DNase I-like superfamily of enzymes that require divalent cation(s) to catalyze phosphoryl-transfer reactions. A new 1.92 Å resolution crystal structure of APE1 reveals ideal octahedral coordination of a single Mg2+ ion and informs on the role of this essential cofactor.
PDB reference: APE1, 4lnd

Download citation


Download citation
Open access
access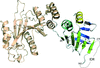
Crystal and small-angle X-ray scattering structures of full-length human SUFU alone and in complex with the conserved SYGHL motif from GLI transcription factors show major conformational changes associated with binding and reveal an intrinsically disordered region crucial for pathway activation.
short communications

Using basic technologies available in any laboratory, a method named `boundary shuffling' has been devised to generate orientated libraries for soluble domain selection.
addenda and errata
Free

Free








