

 journal menu
journal menu





issue contents
December 2014 issue
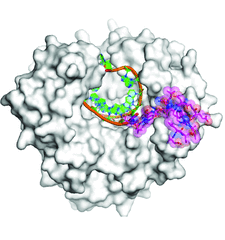
Cover illustration: The overall structure of the RNA-dependent RNA polymerase from norovirus genogroup II (NV RdRP) bound to manganese ions and an RNA primer-template duplex (p. 3099). Residues 5-488 are shown as a solvent-accessible surface and the primer-template RNA duplex is shown in cartoon representation. In this BACK1 complex, residues 489-505 (magenta) are well ordered and are drawn as sticks and as a semi-transparent space-filling representation (magenta).
scientific commentaries





The maximum-likelihood target used for atomic model refinement requires knowledge of statistical parameters. A new method is proposed to estimate them without referring to the test data set thus making refinement more robust.
research papers



Two crystal forms of eIF5B and one crystal form of the eIF5B–eIF1A complex exhibit four different conformations of eIF5B, indicating that the conformation of eIF5B is highly flexible. The interaction between eIF5B domain IV and the eIF1A C-terminal tail may restrict the flexibility of eIF5B on the ribosome and stabilize the interface for ribosome subunit joining.
The crystal structure of a novel human norovirus polymerase replication complex reveals changes in conformation similar to a state following nucleotide incorporation.
PDB reference: human norovirus polymerase replication complex, 4qpx

Evolutionary comparisons suggest that Cwc27 evolved from a prolyl isomerase to a pure proline binder. The increase in the thermal stability of the PPIase domain of Cwc27 from C. thermophilum compared with its human counterpart is based on the additive effects of several amino-acid changes, which result in the removal of long side chains in strained conformations and additional intramolecular interactions.
Open  access
access
access
The maximum-likelihood free-kick target, which calculates model error estimates from the work set and a randomly displaced model, proved superior in the accuracy and consistency of refinement of crystal structures compared with the maximum-likelihood cross-validation target, which calculates error estimates from the test set and the unperturbed model.
Structures of EphA2 representing three activation states reveal that EphA2 probably possesses an alternate activation mechanism distinct from those of other Eph receptor tyrosine kinases.
Open access
access
Crystal structures of the wild type and the N253A mutant of trehalose synthase from D. radiodurans in complex with the inhibitor Tris have been determined at 2.7 and 2.21 Å resolution, respectively, and they display a closed conformation for catalysis of the intramolecular isomerization.
The atomic resolution X-ray diffraction structure of the reduced state of the redox flavoenzyme cholesterol oxidase is presented. The structure indicates the presence of an H atom bound to the flavin N5 atom, suggesting that a hydride transfer has occurred.

This first structural characterization of fully deuterated natural lipid extracts has revealed that the membrane stack is complex and regulated mainly by electrostatic repulsion between adjacent membranes. Structural differences with respect to hydrogenous analogues are investigated as a function of temperature and humidity.
Open access
access
Crystal structures of P. vivax serine hydroxymethyltransferase (PvSHMT) in complex with L-serine and with D-serine and 5-formyltetrahydrofolate provide better understanding of ligand binding and the catalytic mechanism. Features that are important for controlling the activity and specificity of PvSHMT such as stereoselectivity and redox status are addressed.
A conjoined archeal L-asparaginase obtained by assembly of the individual domains in the absence of covalent linker displayed structural similarity and functional superiority than the parent protein. A new mechanism of action has been proposed from the crystal structure of the substrate-bound conjoined enzyme.
Open access
access
The structure of internodal myelin in the rodent central and peripheral nervous systems has been determined using neutron diffraction. The kinetics of water exchange in these tissues is also described.
Structural and enzymological characterization of the human degradative medium-chain-specific 3-ketoacyl-CoA thiolase (hT1) shows that its structure is highly similar to the short-chain-specific tetrameric biosynthetic thiolases, except for small structural differences in two loops at the active site, which provide extra space for a medium-chain fatty-acyl tail to bind. The intrinsic thioesterase activity of hT1 is also discussed.
The structures of retinal-bound and retinol-bound hCRBPII are reported. The X-ray flux-dependent and wavelength-dependent rearrangement of retinol in the binding pocket of hCRBPII is demonstrated. The binding modes of retinol and retinal in CRBPI and CRBPII are contrasted.
The structure of the Tle4-Tli4 complex at 1.75 Å resolution is reported.
PDB reference: Tle4–Tli4 complex, 4r1d
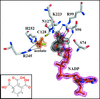
Structure-guided inhibitor development against an essential enzyme in the aspartate-biosynthetic pathway.
PDB references: spASADH–NADP + 4-nitro-2-phosphonobenzoate, 4r41; vcASADH–NADP + 4-nitro-2-phosphonobenzoate, 4r5m; spASADH–NADP + 1,2,3-benzenetricarboxylate, 4r3n; spASADH–NADP + 3-(3-carboxypropyl)phthalate, 4r4j; spASADH–NADP + 3-(3-carboxypropenyl)phthalate, 4r5h; spASADH–NADP + 3-(2-carboxyethyl)phthalate, 4r54; spASADH–NADP + phthalate, 4r51; spASADH–2′,5′-ADP + 1,2,3-benzenetricarboxylate, 4r3w
Open access
access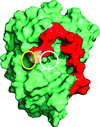
The variable C-terminal tail of manganese peroxidases, a group of enzymes involved in lignin degradation, is implicated in their catalytic and stability properties, as shown by new crystal structures, molecular-simulation and directed-mutagenesis data. Based on this structural–functional evaluation, short and long/extralong manganese peroxidase subfamilies have been accepted; the latter are characterized by exceptional stability, while it is shown for the first time that the former are able to oxidize other substrates at the same site where manganese(II) is oxidized.
The advantages of perdeuteration with regard to solvent visibility in macromolecules is demonstrated through the comparison of two neutron protein structures.
Structural analysis of apo and DNA-bound HU from S. aureus (SHU) revealed that conformational changes occur in both the protein and DNA upon DNA binding. Base-readout and shape-readout mechanisms are involved in DNA binding and recognition by SHU, in which β-arm flexibility is a major determinant.
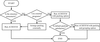
An algorithm is proposed for selecting a unique description of macromolecular crystal structures. It is recommended that authors use the procedure before deposition and publication, so that crystal structures are easier to interpret and compare by noncrystallographers.
Open access
access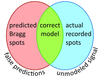
X-ray free-electron laser crystallography relies on the collection of still-shot diffraction patterns. New methods are developed for optimal modeling of the crystals' orientations and mosaic block properties.
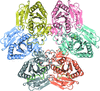
The three-dimensional structures of uridine phosphorylase from S. oneidensis MR-1 in the free form and in complex with uridine were determined at atomic resolution (0.95 Å) and high resolution (1.6 Å), respectively. The uridine molecule has an energetically unfavourable high-syn conformation with a nearly planar ribose ring. The hexamer of uridine phosphorylase was found to be asymmetric.
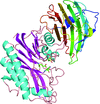
The structure of vanin 1, a human ectoenzyme involved in inflammation and metabolic disease, is shown to be regulated by an unusual allosteric mechanism.







