

 journal menu
journal menu




issue contents
June 2015 issue
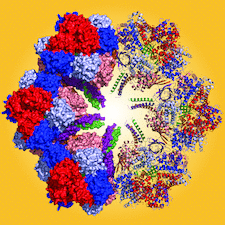
Cover illustration: The giant haemoglobin from Glossoscolex paulistus (Ruggiero Bachega et al., p. 1257). The figure shows how the protomer–protomer interactions occur within the hexagonal monolayer.
scientific commentaries



Free 

Membrane protein structural biology has made tremendous advances over the last decade but there are still many challenges associated with crystallization, data collection and structure determination. Two independent groups, Axford et al. [(2015), Acta Cryst. D71, 1228–1237] and Huang et al. [(2015), Acta Cryst. D71, 1238–1256], have published methods that make a major contribution to addressing these challenges.
research papers


Open access
access
The X-ray structure determination of an integral membrane protein using synchrotron diffraction data measured in situ at room temperature is demonstrated.
PDB reference: HiTehA, 4ycr

Download citation


Download citation
Open access
access
A method for performing high-throughput in situ serial X-ray crystallography with soluble and membrane proteins in the lipid cubic phase is described. It works with microgram quantities of protein and lipid (and ligand when present) and is compatible with the most demanding sulfur SAD phasing.

The structure of the giant haemoglobin from G. paulistus was determined.
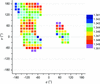
Statistical surveys of protein structures combined with quantum-mechanical calculations reveal a systematic dependence of bond distances on conformation. Interestingly, the variability of backbone C—O and C—N distances is not directly related to peptide-bond planarity.
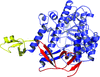
The crystal structure of fission yeast Dis3l2 displays an open conformation that differs from that in the mouse Dis3l2–RNA complex.
PDB reference: Dis3l2, 4ro1

S. aureus steals iron in the form of haem from human haemoglobin (Hb). The crystal structure of the S. aureus Hb receptor IsdH in complex with Hb shows that the receptor induces a conformational change in the globin haem pocket that may promote haem transfer.
PDB reference: IsdH, complex with Hb, 4xs0
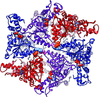
The hexameric structure of truncated A. aeolicus FtsH shows a molecular architecture that forms two rings, in which the ATPase ring possesses twofold symmetry and the protease ring possesses sixfold symmetry. The importance of a dynamic β-strand in the active site and a conserved glycine in the linker region is demonstrated in the full-length protein by site-directed mutagenesis.
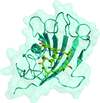
Reported here are two crystallographic structures of rapamycin in complex with the FK506-binding domain of Plasmodium falciparum PfFKBP35 at high resolution, in both its oxidized and reduced forms.
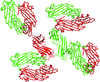
The crystal structure of the μ homology domain of the δ subunit of the bovine COPI complex was determined to 2.15 Å resolution. The structure reveals important aspects of interdomain flexibility that relate to the function of COPI.
PDB reference: MHD of the COPI δ subunit, 4o8q
Open access
access
Crystal structures of the GH130 enzyme Uhgb_MP in the apo form and in complex with mannose and N-acetylglucosamine are described and the structural determinants of the functional specificities of the enzymes involved in N-glycan breakdown by human gut bacteria are identified.
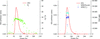
DATASW, a novel tool for the rapid processing of HPLC–SAXS data, is described.
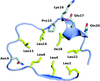
The crystal structures of four representative LRR proteins from the pathogen Leptospira interrogans have been solved, providing insights into a novel subfamily of bacterial LRR characterized by consecutive stretches of a 23 amino-acid repeat motif.
Open access
access
3-Sulfinopropionyl-coenzyme A desulfinase (AcdDPN7) is a novel desulfinase which catalyzes sulfur abstraction in the catabolic pathway of 3,3′-dithiodipropionic acid. The crystal structures of native AcdDPN7 at 1.89 Å resolution and of native AcdDPN7 soaked with the substrate analogue succinyl-CoA at 2.30 Å resolution revealed an acyl-CoA dehydrogenase fold with Arg84 as a key residue in the desulfination reaction.
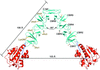
The full-length structure of the major autolysin LytA from S. pneumoniae combined with enzymatic activity assays suggest that dimerization and full occupancy of all choline-binding sites are indispensable for the full activity of LytA in vivo.
PDB reference: LytA, 4x36
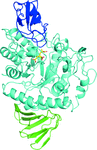
The structures of complexes of HaG and its mutants revealed the substrate specificity and catalytic mechanism of HaG.
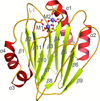
The PPM II phosphatase RsbX features a catalytic centre that binds two Mn2+ ions using five residues instead of the usual four as in PPM I phosphatases, with a conformational reorganization that tilts a flexible loop towards the second Mn2+ ion for coordination of the fifth residue.
Open access
access
An updated partiality model and post-refinement algorithm for XFEL snapshot diffraction data is presented and confirmed by observing anomalous density for S atoms at an X-ray wavelength of 1.3 Å.
PDB reference: 1.46 Å resolution XFEL structure of CPV17, 4zqx







