journal menu
journal menu





issue contents
February 2017 issue
Protein-ligand complexes: understanding biological chemistry (Part 1)
Proceedings of the CCP4 Study Weekend edited by Charles Ballard, Judit Debreczeni and Paul Emsley
Cover illustration: Coot representations of ligand CQ8 in the PDB entry 4zzn.
introduction




Open  access
access
access
An introduction to the Proceedings of the 2016 CCP4 Study Weekend on protein-ligand complex structures.
research papers
Open access
access
This article aims to guide efforts in protein–ligand complex crystal structure generation, with special consideration of protein construct design, and summarizes different approaches to co-crystallization and crystal soaking. Common problems and pitfalls are highlighted.

Open access
access
An overview of the process of ligand restraint generation for macromolecular crystallographic refinement is given.
Open access
access
The entries from a freely available small-molecule database, the Crystallography Open Database, have been validated and a reliable subset of molecules has been selected for the extraction of molecular-geometry information. The atom types and corresponding bond and angle classes derived from this database have been subjected to validation, the results of which are used by AceDRG in the derivation of new ligand descriptions.
Open access
access
The program AceDRG generates accurate stereochemical descriptions, and one or more conformations, of a given ligand. The program also analyses entries and extracts local environment-dependent atom types, bonds and angles from the Crystallography Open Database.
Open access
access
Obtaining a restraint dictionary for novel ligands or improving restraints for known ligands can require their manual modification. This can be tedious and error-prone. REEL is a restraints editor that provides quick, easy and accurate development, allowing global changes and fine-tuning of individual restraints.
Open access
access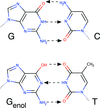
H atoms are `hard to see' in X-ray crystal structures of protein–ligand complexes. This paper discusses the problem of identifying the correct tautomeric form(s) of protein-bound ligands.

Open access
access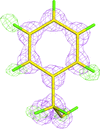
The use of neutron crystallography and in situ spectroscopy to study enzyme mechanism is discussed.
Open access
access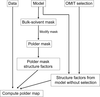
Residual OMIT maps can be improved by the selective exclusion of bulk solvent from the OMIT region.
Open access
access
The process of ligand fitting with CCP4 is reviewed, including identifying ligand density in the map, ligand fitting, refinement and subsequent validation. Recent developments are discussed, and are illustrated using instructive examples demonstrating practical application.
Open access
access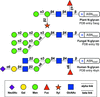
This article addresses many of the typical difficulties that a structural biologist may face when dealing with carbohydrates, with an emphasis on problem solving in the resolution range where X-ray crystallography and cryo-electron microscopy are expected to overlap in the next decade.

Open access
access
With structural glycoscience finally gaining popularity, the need for a clear way of depicting glycans and their interactions in three dimensions is more pressing than ever. Here the Glycoblocks representation is introduced, which combines a simplified bonding-network depiction with the familiar two-dimensional glycan notation brought into three-dimensions.