The crystal structure of the title compound, [Cd(C
4H
10N
4)
3](ClO
4)
2, has been precisely determined at
ca 110 K. The cation is located on a
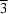
axis and is characterized by an approximate octahedral geometry, with each of the ligands occupying two coordination sites around the metal.
Supporting information
CCDC reference: 269415
Key indicators
- Single-crystal X-ray study
- T = 110 K
- R factor = 0.032
- wR factor = 0.090
- Data-to-parameter ratio = 14.5
checkCIF/PLATON results
No syntax errors found
 Alert level A
CHEM001_ALERT_1_A _chemical_formula_sum is missing
Chemical formula as sum of elements.
The following tests will not be performed.
CELLZ_01,CHEMS_01,CHEMW_01
ABSMU01_ALERT_1_A The ratio of given/expected absorption coefficient lies
outside the range 0.90 <> 1.10
Calculated value of mu = 0.000
Value of mu given = 1.181
DIFMN02_ALERT_2_A The minimum difference density is < -0.1*ZMAX*2.00
_refine_diff_density_min given = -0.584
Test value = 0.000
DIFMX01_ALERT_2_A The maximum difference density is > 0.1*ZMAX*2.00
_refine_diff_density_max given = 0.367
Test value = 0.000
PLAT029_ALERT_3_A _diffrn_measured_fraction_theta_full Low ....... 0.90
Alert level A
CHEM001_ALERT_1_A _chemical_formula_sum is missing
Chemical formula as sum of elements.
The following tests will not be performed.
CELLZ_01,CHEMS_01,CHEMW_01
ABSMU01_ALERT_1_A The ratio of given/expected absorption coefficient lies
outside the range 0.90 <> 1.10
Calculated value of mu = 0.000
Value of mu given = 1.181
DIFMN02_ALERT_2_A The minimum difference density is < -0.1*ZMAX*2.00
_refine_diff_density_min given = -0.584
Test value = 0.000
DIFMX01_ALERT_2_A The maximum difference density is > 0.1*ZMAX*2.00
_refine_diff_density_max given = 0.367
Test value = 0.000
PLAT029_ALERT_3_A _diffrn_measured_fraction_theta_full Low ....... 0.90
| Author Response: The structure of complex (I) is isomorphous and isometric with
the structure of an analogous zinc complex described in a preceeding paper
and analyzed at identical experimental conditions.
Due to technical problems and to save time, a less complete
data set was used in the crystallographic refinement of this structure
without affecting significantly the precision of the structure
determination. Also, the higher angle diffraction in this case has limited
influence on the refined parameters due to the presence of a strong
diffractor (Cd-atom) which dominates the diffraction pattern.
|
PLAT047_ALERT_1_A SumFormula Not Given ........................... ?
 Alert level C
DIFMN03_ALERT_1_C The minimum difference density is < -0.1*ZMAX*0.75
The relevant atom site should be identified.
DIFMX02_ALERT_1_C The minimum difference density is > 0.1*ZMAX*0.75
The relevant atom site should be identified.
REFLT03_ALERT_3_C Reflection count < 95% complete
From the CIF: _diffrn_reflns_theta_max 26.96
From the CIF: _diffrn_reflns_theta_full 0.00
From the CIF: _reflns_number_total 810
TEST2: Reflns within _diffrn_reflns_theta_max
Count of symmetry unique reflns 893
Completeness (_total/calc) 90.71%
PLAT022_ALERT_3_C Ratio Unique / Expected Reflections too Low .... 0.91
PLAT042_ALERT_1_C Calc. and Rep. MoietyFormula Strings Differ .... ?
PLAT420_ALERT_2_C D-H Without Acceptor N2 - H2B ... ?
Alert level C
DIFMN03_ALERT_1_C The minimum difference density is < -0.1*ZMAX*0.75
The relevant atom site should be identified.
DIFMX02_ALERT_1_C The minimum difference density is > 0.1*ZMAX*0.75
The relevant atom site should be identified.
REFLT03_ALERT_3_C Reflection count < 95% complete
From the CIF: _diffrn_reflns_theta_max 26.96
From the CIF: _diffrn_reflns_theta_full 0.00
From the CIF: _reflns_number_total 810
TEST2: Reflns within _diffrn_reflns_theta_max
Count of symmetry unique reflns 893
Completeness (_total/calc) 90.71%
PLAT022_ALERT_3_C Ratio Unique / Expected Reflections too Low .... 0.91
PLAT042_ALERT_1_C Calc. and Rep. MoietyFormula Strings Differ .... ?
PLAT420_ALERT_2_C D-H Without Acceptor N2 - H2B ... ?
 Alert level G
CHEMS02_ALERT_1_G Please check that you have entered the correct
_publ_requested_category classification of your compound;
FI or CI or EI for inorganic; FM or CM or EM for metal-organic;
FO or CO or EO for organic.
From the CIF: _publ_requested_category EM
From the CIF: _chemical_formula_sum :
REFLT03_ALERT_1_G ALERT: Expected hkl max differ from CIF values
From the CIF: _diffrn_reflns_theta_max 26.96
From the CIF: _reflns_number_total 810
From the CIF: _diffrn_reflns_limit_ max hkl 12. 10. 15.
From the CIF: _diffrn_reflns_limit_ min hkl -12. -10. -15.
TEST1: Expected hkl limits for theta max
Calculated maximum hkl 12. 12. 19.
Calculated minimum hkl -12. -12. -19.
Alert level G
CHEMS02_ALERT_1_G Please check that you have entered the correct
_publ_requested_category classification of your compound;
FI or CI or EI for inorganic; FM or CM or EM for metal-organic;
FO or CO or EO for organic.
From the CIF: _publ_requested_category EM
From the CIF: _chemical_formula_sum :
REFLT03_ALERT_1_G ALERT: Expected hkl max differ from CIF values
From the CIF: _diffrn_reflns_theta_max 26.96
From the CIF: _reflns_number_total 810
From the CIF: _diffrn_reflns_limit_ max hkl 12. 10. 15.
From the CIF: _diffrn_reflns_limit_ min hkl -12. -10. -15.
TEST1: Expected hkl limits for theta max
Calculated maximum hkl 12. 12. 19.
Calculated minimum hkl -12. -12. -19.
6 ALERT level A = In general: serious problem
0 ALERT level B = Potentially serious problem
6 ALERT level C = Check and explain
2 ALERT level G = General alerts; check
8 ALERT type 1 CIF construction/syntax error, inconsistent or missing data
3 ALERT type 2 Indicator that the structure model may be wrong or deficient
3 ALERT type 3 Indicator that the structure quality may be low
0 ALERT type 4 Improvement, methodology, query or suggestion
Data collection: COLLECT (Nonius, 1999); cell refinement: DENZO (Otwinowski & Minor, 1997); data reduction: DENZO; program(s) used to solve structure: SIR92 (Altomare et al., 1994); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics: ORTEPIII (Burnett & Johnson, 1996); software used to prepare material for publication: SHELXL97.
Tris(biacetyl dihydrazone-
κ2N,
N')cadmium(II) bis(perchlorate)
at 110 K
top
Crystal data top
| [Cd(C4H10N4)3](ClO4)2 | Dx = 1.783 Mg m−3 |
| Mr = 653.78 | Mo Kα radiation, λ = 0.71073 Å |
| Trigonal, P3c1 | Cell parameters from 1745 reflections |
| Hall symbol: -P 3 2"c | θ = 2.5–27.0° |
| a = 9.5905 (7) Å | µ = 1.18 mm−1 |
| c = 15.2874 (8) Å | T = 110 K |
| V = 1217.72 (14) Å3 | Prism, colourless |
| Z = 2 | 0.20 × 0.15 × 0.15 mm |
| F(000) = 664 | |
Data collection top
Nonius KappaCCD
diffractometer | 810 independent reflections |
| Radiation source: fine-focus sealed tube | 638 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.041 |
| Detector resolution: 56 microns pixels mm-1 | θmax = 27.0°, θmin = 2.5° |
| φ and ω scans | h = −12→12 |
Absorption correction: multi-scan
(Blessing, 1995) | k = −10→10 |
| Tmin = 0.798, Tmax = 0.843 | l = −15→15 |
| 8975 measured reflections | |
Refinement top
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.032 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.090 | H-atom parameters constrained |
| S = 1.08 | w = 1/[σ2(Fo2) + (0.0529P)2 + 0.0248P]
where P = (Fo2 + 2Fc2)/3 |
| 810 reflections | (Δ/σ)max = 0.013 |
| 56 parameters | Δρmax = 0.37 e Å−3 |
| 0 restraints | Δρmin = −0.58 e Å−3 |
Special details top
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes)
are estimated using the full covariance matrix. The cell e.s.d.'s are taken
into account individually in the estimation of e.s.d.'s in distances, angles
and torsion angles; correlations between e.s.d.'s in cell parameters are only
used when they are defined by crystal symmetry. An approximate (isotropic)
treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s.
planes. |
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor
wR and goodness of fit S are based on F2, conventional
R-factors R are based on F, with F set to zero for
negative F2. The threshold expression of F2 >
σ(F2) is used only for calculating R-factors(gt) etc.
and is not relevant to the choice of reflections for refinement.
R-factors based on F2 are statistically about twice as large
as those based on F, and R- factors based on ALL data will be
even larger. The metal cation is located on 3 bar axis, while the perchlorate anion on a 3
rotation axis. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| Cd1 | 0.0000 | 0.0000 | 0.2500 | 0.0190 (2) | |
| N2 | −0.1318 (3) | 0.1119 (3) | 0.08418 (15) | 0.0258 (6) | |
| H2A | −0.1733 | 0.1662 | 0.0617 | 0.029 (9)* | |
| H2B | −0.0233 | 0.1724 | 0.0769 | 0.035 (9)* | |
| N3 | −0.1685 (3) | 0.0606 (3) | 0.16943 (13) | 0.0218 (5) | |
| C4 | −0.3019 (3) | 0.0333 (3) | 0.20477 (17) | 0.0204 (6) | |
| C5 | −0.4208 (4) | 0.0670 (4) | 0.15942 (19) | 0.0269 (6) | |
| H5A | −0.4018 | 0.1735 | 0.1769 | 0.040* | |
| H5B | −0.5304 | −0.0151 | 0.1758 | 0.040* | |
| H5C | −0.4077 | 0.0648 | 0.0959 | 0.040* | |
| Cl6 | 0.6667 | 0.3333 | 0.42187 (8) | 0.0240 (3) | |
| O7 | 0.5030 (3) | 0.2438 (3) | 0.45279 (16) | 0.0389 (6) | |
| O8 | 0.6667 | 0.3333 | 0.3284 (2) | 0.0269 (8) | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| Cd1 | 0.0197 (2) | 0.0197 (2) | 0.0175 (4) | 0.00986 (12) | 0.000 | 0.000 |
| N2 | 0.0278 (14) | 0.0313 (14) | 0.0201 (15) | 0.0162 (12) | 0.0018 (9) | 0.0067 (9) |
| N3 | 0.0238 (13) | 0.0232 (12) | 0.0184 (12) | 0.0117 (10) | 0.0001 (10) | 0.0024 (9) |
| C4 | 0.0204 (14) | 0.0157 (13) | 0.0233 (14) | 0.0077 (12) | −0.0039 (11) | −0.0031 (10) |
| C5 | 0.0255 (15) | 0.0278 (16) | 0.0297 (17) | 0.0149 (14) | −0.0038 (12) | 0.0011 (12) |
| Cl6 | 0.0271 (5) | 0.0271 (5) | 0.0178 (9) | 0.0136 (2) | 0.000 | 0.000 |
| O7 | 0.0357 (14) | 0.0431 (14) | 0.0344 (15) | 0.0171 (11) | 0.0140 (10) | 0.0084 (9) |
| O8 | 0.0313 (12) | 0.0313 (12) | 0.018 (2) | 0.0157 (6) | 0.000 | 0.000 |
Geometric parameters (Å, º) top
| Cd1—N3i | 2.325 (2) | C4—C4ii | 1.489 (5) |
| Cd1—N3ii | 2.325 (2) | C4—C5 | 1.502 (4) |
| Cd1—N3iii | 2.325 (2) | C5—H5A | 0.98 |
| Cd1—N3iv | 2.325 (2) | C5—H5B | 0.98 |
| Cd1—N3 | 2.325 (2) | C5—H5C | 0.98 |
| Cd1—N3v | 2.325 (2) | Cl6—O8 | 1.429 (4) |
| N2—N3 | 1.375 (3) | Cl6—O7vi | 1.441 (2) |
| N2—H2A | 0.87 | Cl6—O7vii | 1.441 (2) |
| N2—H2B | 0.91 | Cl6—O7 | 1.441 (2) |
| N3—C4 | 1.289 (3) | | |
| | | |
| N3i—Cd1—N3ii | 94.53 (7) | C4—N3—N2 | 121.4 (2) |
| N3i—Cd1—N3iii | 94.53 (7) | C4—N3—Cd1 | 118.05 (17) |
| N3ii—Cd1—N3iii | 94.53 (7) | N2—N3—Cd1 | 120.39 (17) |
| N3i—Cd1—N3iv | 106.63 (12) | N3—C4—C4ii | 117.03 (15) |
| N3ii—Cd1—N3iv | 154.27 (12) | N3—C4—C5 | 123.1 (2) |
| N3iii—Cd1—N3iv | 69.82 (11) | C4ii—C4—C5 | 119.87 (16) |
| N3i—Cd1—N3 | 154.27 (12) | C4—C5—H5A | 109.5 |
| N3ii—Cd1—N3 | 69.82 (11) | C4—C5—H5B | 109.5 |
| N3iii—Cd1—N3 | 106.63 (12) | H5A—C5—H5B | 109.5 |
| N3iv—Cd1—N3 | 94.53 (7) | C4—C5—H5C | 109.5 |
| N3i—Cd1—N3v | 69.82 (11) | H5A—C5—H5C | 109.5 |
| N3ii—Cd1—N3v | 106.63 (12) | H5B—C5—H5C | 109.5 |
| N3iii—Cd1—N3v | 154.27 (12) | O8—Cl6—O7vi | 109.14 (12) |
| N3iv—Cd1—N3v | 94.53 (7) | O8—Cl6—O7vii | 109.14 (12) |
| N3—Cd1—N3v | 94.53 (7) | O7vi—Cl6—O7vii | 109.80 (12) |
| N3—N2—H2A | 118.4 | O8—Cl6—O7 | 109.14 (12) |
| N3—N2—H2B | 110.6 | O7vi—Cl6—O7 | 109.80 (12) |
| H2A—N2—H2B | 106.8 | O7vii—Cl6—O7 | 109.79 (12) |
| | | |
| N3i—Cd1—N3—C4 | 55.04 (19) | N3iii—Cd1—N3—N2 | 96.0 (2) |
| N3ii—Cd1—N3—C4 | −0.29 (14) | N3iv—Cd1—N3—N2 | 25.73 (18) |
| N3iii—Cd1—N3—C4 | −89.0 (2) | N3v—Cd1—N3—N2 | −69.19 (15) |
| N3iv—Cd1—N3—C4 | −159.3 (2) | N2—N3—C4—C4ii | 175.7 (3) |
| N3v—Cd1—N3—C4 | 105.8 (2) | Cd1—N3—C4—C4ii | 0.8 (4) |
| N3i—Cd1—N3—N2 | −119.9 (2) | N2—N3—C4—C5 | −5.0 (4) |
| N3ii—Cd1—N3—N2 | −175.3 (3) | Cd1—N3—C4—C5 | −179.9 (2) |
| Symmetry codes: (i) y, x, −z+1/2; (ii) x−y, −y, −z+1/2; (iii) −x, −x+y, −z+1/2; (iv) −x+y, −x, z; (v) −y, x−y, z; (vi) −y+1, x−y, z; (vii) −x+y+1, −x+1, z. |
Comparison of some bond lengths (Å) in the isostructural Cd, Zn and Ni
1:3 complexes with biacetyldihydrazone. top| Bond | M = Zn | M = Cd | M = Ni |
| | | |
| M—N3 | 2.158 (2) | 2.325 (2) | 2.060–2.088 |
| N2—N3 | 1.386 (3) | 1.375 (3) | 1.375–1.388 |
| N3—C4 | 1.281 (3) | 1.289 (3) | 1.278–1.298 |
| C4—C5 | 1.492 (3) | 1.502 (4) | 1.378–1.504 |
| C4—C4* | 1.502 (5) | 1.489 (5) | 1.473–1.485 |


 journal menu
journal menu metal-organic compounds
metal-organic compounds













 Alert level A
Alert level A
 Alert level C
Alert level C
 Alert level G
Alert level G



