journal menu
journal menu




issue contents
June 1998 issue
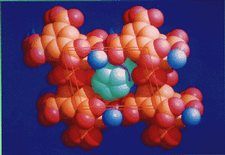
Cover illustration: Space filling representation of the hydrated anionic framework formed by trimesic acid around piles of cobalticinium cations in crystalline [(C5H5)2Co][(H3TMA)(H2TMA)].2H2O. Graphics (picture or drawing) obtained using SCHAKAL by E. Keller, University of Freiburg, Germany. Courtesy of D. Braga and F. Grepioni
research papers



Download citation





Download citation

X-ray diffraction and Raman spectroscopy have shown that the crystal structure of Cs2HgBr4 in the normal, incommensurate and low-temperature phases can be described by disordered models.

The bond-valence and resonance-bond-number methods for calculating interatomic distances are compared for KVO3, β-Ga2O3, TeI4, Li2SiO3, Li2GeO3 and CaCrF5.
Download citation
Download citation

Hexagonal (Na3/4K1/4)AlGeO4 is isostructural with the mineral nepheline, (Na3/4K1/4)AlSiO4, with a similar split oxygen position and a cation deficiency on the potassium site. In contrast, the alkali sites are fully occupied in the structure of monoclinic (Na3/4K1/4)AlGeO4 and the lower unit-cell symmetry eliminates the oxygen disorder.

The maximum entropy method provides a means of ranking bond graphs in order of symmetry and gives good predictions of the bond valences.

Comparison of Experimental and Theoretical Structure Amplitudes and Valence Charge Densities of GaAs
The valence and difference charge densities of GaAs obtained from experimental and theoretical data (Hartree–Fock and density functional theory) show a charge accumulation between nearest neighbours slightly shifted towards the arsenic site. The disagreement remaining between experimental and theoretical data and between those of both ab initio methods are of the same order of magnitude.

Distinct types of diffuse scattering which appear in the electron diffraction patterns of tetragonal tungsten bronze-type (TTB) niobium tungsten oxides are caused by different arrangements of filled pentagonal tunnels. A structural model for a new TTB superstructure (Nb6W8O39) has been derived from HRTEM images.
Download citation
Download citation

A new classification based on the geometry of the coordination polyhedron, especially useful for six-coordinate bis(salicyl- aldiminato) complexes, has been proposed. The distortions of octahedral Ni2+ complexes have been analyzed and exemplified by a crystal structure of a complex showing the largest shift of the metal from the geometric center observed so far in similar complexes.
CCDC reference: 131977
Download citation
Download citation

The oxalic acid complexes of DL and L forms of basic amino acids exhibit a variety of ionization states and stoichiometries. They demonstrate the tendency of individual molecules to retain their intrinsic aggregation propensities even as they interact with other molecules.
Download citation
Download citation

Structures of the cyclic derivatives of o-acylbenzoic acids, including the chloride, endo- and exocyclic amides, the ester and two types anhydride, are described. Former aldehyde/ketone carbon–heteroatom endocyclic and exocyclic bond distances show length variations which correlate with the relative basicities of the attached groups.
Download citation
Download citation

Physico-chemical studies of the phase transformations by Chattaway and co-workers (1911–1915) are extended by structure determinations for the four phases involved and DSC measurements on the transformations.

An attempt has been made to unify and extend previous predictions of the possible crystal packings of benzene. Dozens of hypothetical structures were generated, but the two experimentally observed ones still have the lowest enthalpy at their respective pressures.
Download citation
Download citation

Z-(Aib)9OBut contains the unusual α-aminoisobutyric acid. The structure of this synthetic oligopeptide reveals a rare C-terminal conformation in the form of a disorder of one of the two molecules in the asymmetric unit.
CCDC reference: 131984
Download citation
Download citation

The precision of previous studies may have been limited by twinning. Analyses of data collected at five temperatures show that the temperature dependence of the rigid-body motion is normal and confirm that the out-of-plane motion of the O atoms is large.

A re-investigation of the formation of hydrogen bonds to the thiocyanate anion demonstrates that preferred hydrogen-bond geometries are in the directions of the sulfur and nitrogen lone pairs of the anion.

Three C=O⋯C=O interaction motifs are commonly observed in crystal structures: the antiparallel, perpendicular and sheared-parallel motifs. Ab-initio molecular-orbital calculations on a bis-propanone dimer model yield interaction energies of -22 kJ mol−1 for the antiparallel motif, having two short C⋯O interactions, and -7.6 kJ mol−1 for the perpendicular motif, having only one such short dipolar interaction.
Download citation
Download citation

Pairwise-interwoven nets built from tris-phenol molecules are cross-linked into two-dimensional bilayers by 1,2-diamino- ethane molecules.
CCDC reference: 131963
Download citation
Download citation

Refinement of uric acid dihydrate as both twinned and disordered in space group P21/c fully accounts for inconsistencies present in previous orthorhombic descriptions.
CCDC reference: 131964

Five proposed structures are compared with the reduced density function derived from neutron diffraction data. A T symmetry structure provides the best agreement.