

 journal menu
journal menu




issue contents
June 1999 issue
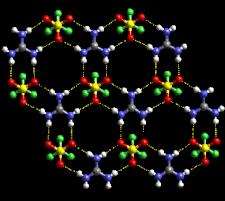
Cover illustration: An infinite hydrogen-bonded sheet in guanidinium trifluoromethanesulfonate. Courtesy of C. B. Aakeröy [Acta Cryst. (1997), B53, 569-586; structure determined by V. A. Russell, M. C. Etter & M. D. Ward].
research papers



The lattice dynamics of Na4TiP2O9 (NTP) and Na4.5FeP2O8(O,F) (NFP) crystals, which are superionic conductors with Na+-ion conductivity, were studied under high pressures. Lattice constants as functions of hydrostatic pressure were measured on a four-circle diffractometer using a high-pressure cell with diamond anvils.

Download citation




Download citation
Investigation of the cation distributions in Y3Al5−xGaxO12 (0 ≤ x ≤ 5) garnet solid solutions using single-crystal X-ray diffraction shows that Ga3+, which is larger than Al3+, preferentially occupies the tetrahedral site rather than the octahedral site, contrary to the effect of cation size. This peculiar cation distribution is most probably caused by both the strong covalency of the Ga—O bond and the need to decrease the neighbouring cation–cation repulsive force.
Download citation
Download citation
The experimental electron density features derived from both high-resolution Mo Kα and Ag Kα X-ray diffraction data are compared for the natural mineral α-spodumene, LiAl(SiO3)2. The kappa-refinement atomic net charges have been used to calculate the Madelung potential. An estimate of the chemical formula electrostatic energy is given.
Download citation
Download citation
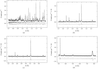
The structure of β-Cs3(HSO4)2[H2−x(SxP1−x)O4] has been studied by neutron diffraction methods and can be adequately described by two different models for the disorder on H and S/P atom sites. The disorder is responsible for the relatively high proton conductivity at room temperature.
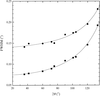
On the basis of microstructure characteristics (strain and crystallite size) and lattice parameters investigated by X-ray powder diffraction methods, a structural model for the SiC–C solid solution is proposed. The behaviour of the structure under high-pressure and high-temperature sintering and annealing in a vacuum is described.
Download citation
Download citation
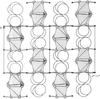
The (isomorphous) structures of Bi14WO24 and Bi14MoO24 have been solved and refined using neutron powder diffraction data in the space group I4/m. The structure adheres closely to a fluorite-type δ-Bi2O3-related parent structure. The metal atoms are compositionally ordered. Substantial displacive disorder is evident around the W site, which appears to be coordinated by a highly disordered O-atom tetrahedron.
Download citation
Download citation
The crystal structure of Ca7Zr7Ta6O36 is solved and refined in space group Fddd using single-crystal X-ray diffraction data.
Download citation
Download citation
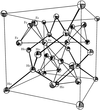
The deformation electron density (Δρ) for the HoFe2 Laves compound obtained from synchrotron X-ray diffraction data shows excess electron density midway along the Ho–Ho, Ho–Fe and Fe–Fe contacts which challenges the conventional understanding of electron interactions in magnetic materials.
Download citation
Download citation
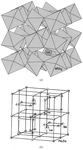
A five-wavelength MAD experiment conducted at ELETTRA Sincrotone, and subsequent crystallographic analysis, shows that substitution of cobalt in CoZnPO-CZP occurs primarily at one of the two sites, with an occupancy of 24–30%. Site-specific occupancies of as low as 12–15% should, in general, be discernible by this method.
Download citation
Download citation
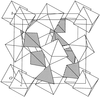
The structure of the negative-thermal-expansion material ZrW2O8 has been determined by parametric Rietveld refinement of high-resolution neutron powder diffraction data collected at 260 temperatures between 2 and 520 K. Despite data collection times of just 5 min at each temperature, it is shown that full anisotropic refinement of the structure is possible.
Download citation
Download citation
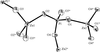
Disparate atomic displacements in skutterudite-type LaFe3CoSb12, a model for thermoelectric behavior
Mean-square atomic displacements in LaFe3CoSb12 show that the La atom exhibits an anomalously large displacement. The large-amplitude rattling of the La atom is linked to the dramatic decrease of the lattice thermal conductivity and this structure–property relationship offers a new paradigm for the exploration of thermoelectric materials.
Download citation
Download citation
The room-temperature crystal structure of Sr(Yb0.5Nb0.5)O3 is monoclinic and can be derived from a triclinic distortion of the prototype cubic cell. The ordering of the different species of B-site cation, Yb3+ and Nb5+, inherently causes a volume difference between the YbO6 and NbO6 octahedra. The triclinic distortion of the prototype cell is caused by a−a−c+ octahedral tilting of the two octahedra with differing volumes. This volume difference is shown to inevitably inhibit the rigid rotation of the octahedra.

The crystal structure of Ga2O3(ZnO)m studied by high-resolution analytical transmission electron microscopy is reported.
Download citation
Download citation
Accurate very low temperature synchrotron-radiation data can now be measured within days, making electron-density studies of compounds beyond the first transition series more frequently within reach.
Download citation
Download citation
Thermal vibration analysis indicates that an experimentally determined distance between atomic centroids can be significantly different from the mean interatomic distance that is of chemical interest. This is especially true for distances involved in weak interactions.
Download citation
Download citation
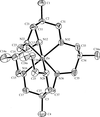
These isomorphic crystal structures are described as intergrowths of four substructures, each a different modulation of an idealized parent structure of space group C2/c. Two substructures correspond to alternative orientations of a C1¯ structure for which x = 0, and two substructures correspond to alternative origins of a P21/n structure for which x = 1. The MN6 coordination geometry is essentially octahedral, in contrast to the trigonal-prismatic geometry observed for the CdII and HgII complexes of the same ligand.
Download citation
Download citation
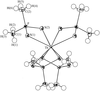
The crystal structures of the three supercomplexes [18]aneN6H2K[Co(CN)6].4H2O, [16]aneN4H2K[Co(CN)6] and [12]aneN4H3[Co(CN)6].2H2O, and the second-sphere interactions in the binding of the [Co(CN)6]3− complex ions with the protonated polyammonium macrocycles are reported.
Download citation
Download citation

The chloromercurate(II) salts of monochloropyridines display a variety of anion stoichiometries and structures. The 2-chloropyridinium salt has the anion stoichiometry [HgCl3]−, but the anions are infinite chains composed of [HgCl3]−, HgCl2 and Cl− moieties linked by longer Hg⋯Cl contacts. Chains of interconnected [HgCl4]2− and HgCl2 entities in the 3- and 4-chloropyridinium salts yield the rare [Hg3Cl10]4− stoichiometry. Second forms of the 3- and 4-chloropyridinium salts contain distorted [Hg2Cl6]2− moieties linked into chains and symmetrical discrete [Hg2Cl6]2−anions, respectively.
Download citation
Download citation

The planarity of the 4,8,12-trioxa-4,8,12,12c-tetrahydrodibenzo[cd,mn]pyrenylium cations in several salts and their structural chemistry are described.
Download citation
Download citation
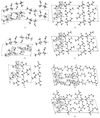
The crystal structures of seven complexes of L-isoleucine with various D-amino acids with hydrophobic side chains are reported. Similarities and differences in the molecular packing and in the conformation of the partner molecule are discussed.
Download citation
Download citation
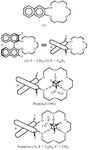
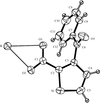
The crystal structure of a highly discriminating chiral host, in its complex with a chiral cationic guest, shows the influence of C—H⋯O and C—H⋯π interactions on the unanticipated orientation of the guest in the host cavity. These interactions are very similar, but not identical, in the two independent complexes in the asymmetric unit.
Download citation
Download citation
The crystal and molecular structure of 3(5),4-dimethylpyrazole and 3,4,5-trimethylpyrazole have been determined at 200 K. Ab initio calculations at HF and B3LYP/6-31G** levels have been undertaken in order to estimate the influence of the methyl substituent on the pyrazole.
Download citation
Download citation

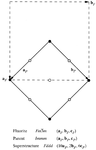
Phase III of the 1:1 adduct of hexamethylenetetramine and azelaic acid presents phase-transition-generated TLQS twinning, with a superposition of a carboxylic acid and a carboxylate form of azelaic acid. Four possible single-domain states are predicted by group theory; only one type of domain wall is observed and is explained in terms of intermolecular interactions.





