

 journal menu
journal menu




issue contents
February 2000 issue
Cover illustration: Perspective view of the structure of the complex of 2,4,6-tris- (4-chlorophenoxy)-1,3,5-triazine with tribromobenzene, determined by neutron diffraction. Courtesy of J. A. K. Howard & C. K. Broder (University of Durham, UK), G. R. Desiraju, A. Nangia & R. K. R. Jetti (University of Hyderabad, India), C. C. Wilson & D. A. Keen (Rutherford Appleton Laboratory, Chilton, UK).
editorial



Free 

research papers

Download citation




Download citation
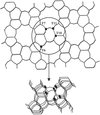
The crystal structures of orthorhombic and monoclinic defective silicalite have been refined from neutron powder diffraction data. Preferential location of Si-atom vacancies was found on four out of 12 independent T sites in the orthorhombic silicalite.
Download citation
Download citation
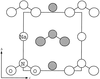
The deformation electron density of NaNO has been determined at 30 K using Hirshfeld deformation functions. Spontaneous polarization was calculated from the results and compared with the measured value.
has been determined at 30 K using Hirshfeld deformation functions. Spontaneous polarization was calculated from the results and compared with the measured value.
Download citation
Download citation

The refinement of the incommensurately modulated structure of Rb2CoBr4 at 295 and 200 K is reported. Results compare well with Rb2ZnBr4.
Download citation
Download citation

Quantum mechanical calculations were performed for two polymorphs of Cu6PbO8. The reconstructive structural phase transition between these two polymorphs is predicted to occur at 18 GPa.
Download citation
Download citation
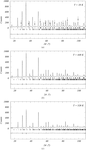
High-resolution neutron powder diffraction data, complemented by those of neutron powder thermodiffractometry, were used to characterize the structural behaviour of the ferroelectric PbHf0.8Ti0.2O3 between 10 and 770 K. Cationic displacements and rotation and/or distortion of oxygen octahedra have been studied through the sequence of phase transitions in order to link structural changes to the electrical properties.
Download citation
Download citation
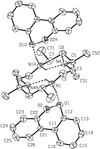
Co-crystallization of meso-5,5,7,12,12,14-hexamethyl-1,4,8,11-tetraazacyclotetradecane with phenols and acids yields salt-like adducts whose hydrogen-bonded supramolecular structures are of varying dimensionality. The adduct with 2,2′-biphenol forms a finite, zero-dimensional aggregate, while those with 4,4′-thiodiphenol, 4,4′-sulfonyldiphenol and 3-hydroxybenzoic acid form one-dimensional molecular ladders; two-dimensional networks are formed in the adducts with 3-hydroxybenzoic acid and phenylphosphonic acid, and three-dimensional frameworks are formed in the adducts with 3,5-dihydroxybenzoic acid and 4,4′-biphenol.
Download citation
Download citation
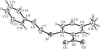
Isolated molecules of 2-nitrophenylthiolates, O2NC6H4SX, exhibit two conformational minima, the global minimum in which the nitro group is coplanar with the adjacent aryl ring and a local minium in which rotation of the substituent X about the exocyclic C—S bond is associated with a disrotatory motion of the nitro group out of the ring plane. In crystalline solids both conformations are observed, but the choice of the local, higher-energy minimum is generally associated with the occurrence of intermolecular C—H⋯O hydrogen bonds.
Download citation
Download citation
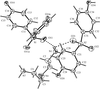
The adducts of bis(2-aminoethyl)amine–4,4′-sulfonyldiphenol (1/3) and bis(2-aminoethyl)amine–1,1,1-tris(4-hydroxyphenyl)ethane (a 1/4/1 methanol solvate) both have structures built from ladders linked into three-dimensional frameworks, while the 1:2 adduct with 3,5-dihydroxybenzoic acid contains interwoven sheets further linked into a three-dimensional framework. The methanol-solvated 1:3 adduct of tris(2-aminoethyl)amine with 4,4′-biphenol can be described in terms of ten-component supermolecules linked into a three-dimensional framework.
Download citation
Download citation
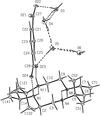
meso-5,5,7,12,12,14-Hexamethyl-1,4,8,11-tetraazacyclotetradecane and terephthalic acid form two different hydrated 1:1 adducts, both of which form chains of alternating [C16H38N4]2+ and [C8H4O4]2− ions linked by N—H⋯O hydrogen bonds. In the tetrahydrate the water molecules form continuous hydrogen-bonded chains which link the ionic chains into a three-dimensional framework; in the hexahydrate, the anions and the water molecules form two-dimensional nets which are linked by the ionic chains into a three-dimensional framework.
Download citation
Download citation
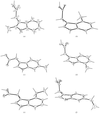
As part of molecular recognition studies on the phytohormone indole-3-acetic acid (IAA) a series of alkylated (alkyl = methyl and ethyl) IAAs has been examined. The structure–activity relationship includes the geometrical parameters of the molecular structures determined by X-ray analysis, electronic properties (from the UV and 1H NMR spectra), growth-promoting activities in the Avena coleoptile straight-growth bioassay and relative lipophilicities. Lipophilicities are correlated with the moments of inertia, average polarizability, molecular mass and the van der Waals radii of the ring substituents.
Download citation
Download citation
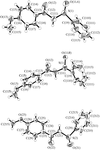
Preferred conformations in the solid state of some α-(p-phenylsulfinyl)-p-substituted acetophenones
The crystal structures of three α-(p-phenylsulfinyl)-p-substituted acetophenones are reported and the stabilization of their conformations in the crystal are discussed in terms of the dipole moment coupling, Coulombic and intramolecular charge transfer interactions between the oppositely charged atoms of the C=O and S=O dipoles.

In multipole refinements of non-centrosymmetric structures based on noisy diffraction data, the errors in the resulting electron density distribution, owing to uncertainty in the phases of the structure factors, do not allow the determination of the interaction density, at least not in the case of urea.
Download citation
Download citation
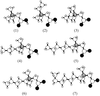
Of the title compounds, those with alkyl chain lengths less than n = 5 form three-dimensional networks with completely different structure. The long-chain compounds form more or less regular bilayers with interdigitated chains.
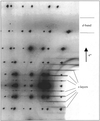
A temperature-dependent quantitative analysis of the widths of diffuse layers in urea inclusion compounds shows that alkane guest molecules adopt a one-dimensional longitudinal paracrystalline arrangement. The degree of ordering increases with decreasing temperatures.
Download citation
Download citation
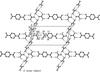
New crystalline adducts of tetraalkylammonium terephthalate/trimesate with urea and water molecules result from hydrogen-bond directed assembly of complementary acceptors and donors that generate anionic channel- and layer-type host lattices for the accommodation of bulky hydrophobic cations.
Download citation
Download citation

The charge density topology of glycyl-L-threonine has been analyzed using single-crystal X-ray diffraction data at 100 K. The experimental electrostatic potential around the molecule is discussed in terms of molecular interactions.
addenda and errata
Free

international union of crystallography
Free





