journal menu
journal menu




issue contents
October 2002 issue
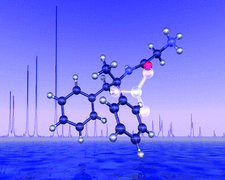
Cover illustration: The structure of remacemide nitrate (C17H21N2O+·NO3-) solved from powder diffraction data using a global optimization approach based on maximum likelihood. Only the remacemide molecule is optimized while the nitrate ion is treated as an unknown blur [Markvardsen, David & Shankland, Acta Cryst. (2002), A58, 316-326]. Image produced by Dr A. Florence, University of Strathclyde, Scotland, using POV-ray 3.1.
research papers



Download citation





Download citation

Three new synthetic langbeinite structures, K2ErTi(PO4)3, K2YbTi(PO4)3 and K2YTi(PO4)3, are presented as well as a reinvestigation of K2CrTi(PO4)3. An alternative approach for the description of cation cages in the langbeinite framework is given using [M5X6O39] building units.
Download citation
Download citation
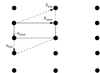
The structure of TlH2PO4 at 180 K has been determined by X-ray diffraction in the space group P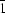 . A comparison with the room-temperature antiferroelectric structure of TlD2PO4 shows that the structural distortions they undergo with respect to the common high-temperature phase are not analogous.
. A comparison with the room-temperature antiferroelectric structure of TlD2PO4 shows that the structural distortions they undergo with respect to the common high-temperature phase are not analogous.
Download citation
Download citation
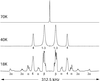
The equilibrium positions of the H atoms in (NH4)2S2O8 are determined using deuteron NMR spectroscopy, mainly at T = 17 K. The results are verified by X-ray diffraction and constitute the background for a discussion of the complex tunnelling and stochastic jump dynamics of the ammoniuum ions.

Bond lengths have been compared in molecules and crystals of alkali halides and alkaline earth metal fluorides and oxides. It was concluded that in alkali halide crystals the counterions considered as compressible spheres do not have common boundaries.
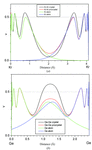
The approximate determination of the localized-orbital locator for detecting the features of chemical bonds in terms of the local kinetic energy is described. Characterization of the covalent, ionic and van der Waals bonds is possible using this approach.
Download citation
Download citation
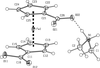
Ferrocene-1,1′-dicarboxylic acid forms adducts with a wide range of organic amines: the structures of nine examples show that most are salts and that hydrogen bonds of O—H⋯O, O—H⋯N and N—H⋯O types can link the components into one-, two- or three-dimensional arrays.
Download citation
Download citation
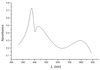
Crystal structure analysis of [(VB1)2DMFHPMo12O40·5DMF] [VB1 = vitamin B1 (thiamine chloride), DMF = N,N-dimethylformamide] indicates that two sets of PO4 tetrahedra are formed around the central P atom. The PO4 tetrahedra and MoO6 octahedra are disordered in the heteropolyanion.
Download citation
Download citation

X-ray single-crystal and powder diffraction and neutron powder diffraction analysis were used to study the structure of BaC2O4·0.5H2O and in situ X-ray powder diffraction was used to study the thermal decomposition of BaC2O4·0.5H2O to α-BaC2O4.
Download citation
Download citation
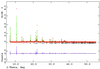
The structure of a series of divalent metal terephthalate salts was determined by applying Monte Carlo simulated annealing techniques to X-ray powder data. The hydrogen bonding was characterized using quantum calculations.
Download citation
Download citation
The crystal structures of three substituted arenesulfonamides have been solved from X-ray powder diffraction data using a new direct-space structure solution method based on a differential evolution algorithm: N—H⋯O hydrogen bonding leads to supramolecular aggregates in three, two or one dimensions, modified in some cases by C—H⋯O hydrogen bonds and aromatic π⋯π stacking interactions.
Download citation
Download citation
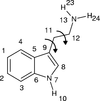
The crystal structure determination of tryptamine free base from powder X-ray diffraction data is described, including an investigation into the effect of the weight of restraints during Rietveld refinement.
Download citation
Download citation

Crystals of CH7N4,ZrF5 subjected to X-radiation exhibit significant variation in axial lengths and decreased standard reflection intensities. Analysis of the variation in atomic coordinates determined on four separate crystals reveals the presence of small but highly significant radiation-induced structural changes.
Download citation
Download citation

Channels in the structure of L-alanyl-L-valine act as supramolecular hosts for organic solvent molecules. The crystals are stable after the complete removal of small guests. Irreversible absorption of alcohols larger than methanol induces a doubling of two axes' lengths and a change of the shape and size of the pores.
Download citation
Download citation
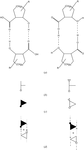
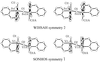
Racemic crystals of three optically active molecules, containing a saturated eight-membered ring bearing two vicinal (either cis or trans) hydrogen-bond donor/acceptor groups, were found to represent two novel forms of supramolecular close packing. The crystals of trans isomers (carboxylic acid and carboxamide) are isostructural, while the cis isomer (carboxylic acid) exhibits only one-dimensional isostructurality with them.
Download citation
Download citation
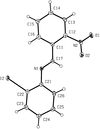
In crystalline nitrobenzylidene-iodoanilines, isolated molecules may occur, or the molecules may be linked by some combination of C—H⋯O hydrogen bonds, iodo⋯nitro interactions and aromatic π⋯π stacking interactions, so giving overall supramolecular structures in zero, one, two or three dimensions.
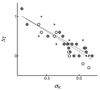
Bond angles and bond lengths in monosubstituted derivatives of benzene and ethene were calculated using the density functional theory. They agree reasonably with available values derived statistically from crystallographic data but correlate poorly with the reactivity parameters.
Download citation
Download citation
Correction of space-group errors for some crystals leads to clarification of relationships among chemically similar but apparently crystallographically different crystal structures. There are new examples of the `centrosymmetric–non-centrosymmetric' ambiguity and also straightforward revisions of space groups. Finally, a questionable structure presents evidence either for a new type of N—H⋯Cl hydrogen bonding or for an error.
Download citation
Download citation

A survey of the October 2001 release of the Cambridge Structural Database has uncovered approximately 675 separate apparently reliable entries under space groups 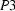 ,
, 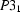 ,
, 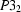 and
and 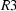 ; in approximately 100 of these entries, the space-group assignment appears to be incorrect. Other features of these space groups are also discussed.
; in approximately 100 of these entries, the space-group assignment appears to be incorrect. Other features of these space groups are also discussed.
short communications

Ab initio calculations on S-alkylated isothiosemicarbazones were performed in order to investigate the role of intra- and intermolecular effects in determining the molecular conformation.