

 journal menu
journal menu




issue contents
June 2009 issue
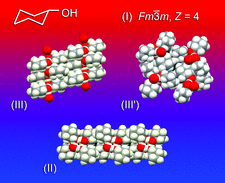
Cover illustration: Though cyclohexanol is a simple compound it has a rich phase diversity at low temperature. It forms a disordered glassy phase on cooling, but this can be transformed into ordered phases (II), (III) and the metastable (III'), which all have different hydrogen-bonding motifs. The series of transitions is attributable to the conformational flexibility of the hydroxyl group [Ibberson et al. (2008). Acta Cryst. B64, 573-582].
feature articles



Download citation





Download citation
Free 
The basic principles of handling and describing modulated structures with the superspace formalism are given in a non-mathematical way. They are illustrated using the crystal structure of (6R,7aS)-6-(tert-butyl-dimethylsilanyloxy)-1-hydroxy-2-phenyl-5,6,7,7a-tetrahydropyrrolizin-3-one, C19H27NO3Si.

Free
This article focuses on weberite ceramics. The structural features of weberite, its relationship to the fluorite and the pyrochlore structures, and some weberite ceramics with interesting properties are discussed.
research papers
Download citation
Download citation

The structures of KxNa1 − xNbO3 (x = 0.05, x = 0.3) at low temperatures have been determined by neutron powder diffraction studies. The phase-coexistence region at the transition between rhombohedral and monoclinic phases was also identified, consistent with a first-order phase boundary.
A number of strict rules are proposed to model the crystal structures of intermetallics as a network of cluster precursors. According to these rules the self-assembly of the ZrZn22-like structures was considered within the hierarchical scheme: primary polyhedral clusters (one AX16 + 2BX12) → zero-dimensional nanocluster precursor (AB2X37) → one-dimensional primary chain → two-dimensional microlayer → three-dimensional microframework (three-dimensional supraprecursor).
Download citation
Download citation

The structures of a family of three tantalum copper aluminides with giant unit cells have been determined. Their mutual relationships are emphasized by a comparison of their average structures.

The complex cluster structures of a family of three tantalum copper aluminides with giant unit cells are discussed.
Download citation
Download citation
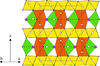
The high-pressure behaviour of the mixed-valence vanadate BaV6O11 was studied with single-crystal X-ray diffraction in a diamond–anvil cell; the pressure-induced transition from P63/mmc to P63mc was studied in detail.
Download citation
Download citation
The crystal structure of Cu2(OH)3Cl has been refined using two nearly stoichiometric natural specimens. The occurrence of a triclinic polymorph is suggested for pure Cu2(OH)3Cl, while clinoatacamite is found for small but significant impurity concentrations.
Download citation
Download citation

For SiBr4 crystal structures were predicted by global lattice-energy minimizations. Two predicted structures could be verified experimentally.
Download citation
Download citation

The structure of racemic [Ca(C4H4O6)]·4H2O [form (II)] is clarified from powder and single-crystal X-ray diffraction data.
Download citation
Download citation
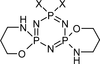
The absolute configurations of pairs of enantiomers separated by high-performance liquid chromatography have been confirmed by single-crystal structure analysis and provide a reference system for subsequent assignment of such systems based on elution order. The solid-state structures of the pairs of enantiomers do not fully adhere to expected enantiomorphous crystallization behaviour.
Download citation
Download citation
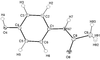
The charge density of the monoclinic paracetamol drug has been derived from high-resolution X-ray diffraction. Topological features of the electron density and electrostatic potential are presented.
Download citation
Download citation
Hantzsch 1,4-dihydropyridine ester analogs, including the prototypical hypertension drug Nifedipine, are of pharmaceutical use but are also good candidates for generating a reproducible packing motif that is characterized by intermolecular N—H⋯O hydrogen-bonded C(6) extended chains.

Simple Monte Carlo models have been developed that reproduce in detail the diffuse diffraction patterns observed for the two room-temperature polymorphs of the drug benzocaine. The analyses reveal highly correlated molecular displacements in both forms, but in polymorph (II) the motions show non-harmonic characteristics that appear to be precursors of a phase transition to a new polymorph which has been found at low temperatures (150 K).
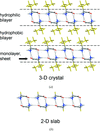
Periodic two-dimensional slabs and full three-dimensional networks were extracted from the X-ray structures of hydrophobic amino acids and then optimized with ab initio methods. The derived interaction energies support the observed propensity for inclusion of two molecules in the asymmetric unit.
addenda and errata
Free

Systematic variations of the bond-valence sums calculated from the poorly determined bond-valence parameters have been illustrated using a simple graphical scheme.
book reviews
Free





