journal menu
journal menu




issue contents
December 2010 issue
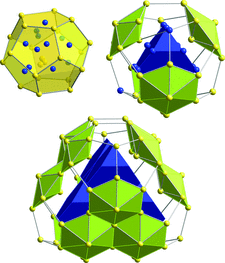
Cover illustration: The clusters shown on the cover image belong to the fundamental structural building units of cF(5928-x)-Al56.6Cu3.9Ta39.5, x = 20, one representative of a family of cluster-based tantalum copper aluminides with giant unit cells hosting up to more than 20 000 atoms. Their structures can be geometrically described as packings of clusters such as fullerenes, dodecahedra, pentagonal bifrusta, hexagonal bipyramids and Friauf polyhedra [Conrad et al. (2009). Acta Cryst. B65, 318-325; see also Weber et al. (2009). Acta Cryst. B65, 308-317].
research papers












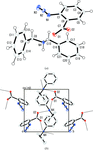
Various reaction pathways of arylnitrenes produced by photo-irradiation in complex crystals composed of 2-, 3- or 4-azidobenzoic acid and a diaryl- or dicyclohexylamine were directly observed by X-ray crystal structure analysis.