

 journal menu
journal menu





issue contents
March 2000 issue

Cover illustration: Conserved side-chain substitutions induce a helical shift in FKBP12.6 (p. 266).
topical reviews




Some of the lessons that were learned during the 1990s with respect to protein model quality and their implications for refinement and rebuilding practices are discussed. In addition, a compendium of criteria that gauge various aspects of the quality of (protein) models is presented.
research papers


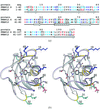
FKBP12.6, a novel isoform fo FKBP12, which selectively binds to the cardiac ryanodine receptor (RyR2) has been crystallized and its structure determined at 2.0 Å resolution.
PDB reference: rapamycin–FKBP12.6 complex, 1c9h
A complex structure between human pepsin, an aspartic proteinase, and a phosphorus-containing inhibitor is presented. The extensive interactions made by this inhibitor enables the characterization of the binding sites from S4 to S3′.
PDB reference: pepsin–inhibitor complex, 1qrp
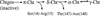
The structure of δ-chymotrypsin bound to a peptidyl inhibitor has been determined at 2.14 Å resolution. This is the first refined structure of δ-chymotrypsin. The structure shows the tetrahedral conformation of the bound substrate analogue and reveals an unusual lattice-disordering effect.
PDB reference: δ-chymotrypsin complex, 1dlk
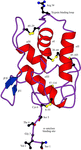
Structure of the bifunctional inhibitor of trypsin and α-amylase from ragi seeds at 2.2 Å resolution
The structure of the bifunctional inhibitor of trypsin and α-amylase has been determined at 2.2 Å resolution. The conformations of the trypsin and α-amylase binding sites are significantly different to those observed in solution by NMR but are very similar to those determined in the crystal structure of the complex between the bifunctional inhibitor and α-amylase.
PDB reference: trypsin/α-amylase inhibitor, 1b1u
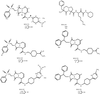
Five new non-electrophilic β-strand-templated thrombin active-site inhibitors are described as they bind to thrombin combining the non-transition state selectivity of DAPA-like inhibitors with the more optimal binding features of the S1–S3 sites of peptidic molecules, which results in highly potent and selective inhibitors of thrombin.

To investigate a potential candidate material for making artificial red blood cells for supplement blood transfusion, the crystal structure of porcine haemoglobin was determined up to 1.8 Å resolution.
PDB reference: porcine haemoglobin, 1qpw
A special type of pseudo-symmetry is described which gives the same amplitude to both pseudo-equivalent reflections, making it impossible to determine the correct point group on the basis of Rmerge.
The quality of electron-density maps derived from a limited number of experimentally determined triplet phases is discussed, as well as some methodological aspects concerning the minimum number of phases needed for a successful structure solution.

Radiation damage, as can occur during data collections from cryo-cooled protein crystals, has been shown to cause the breakage of disulfide bonds and the decarboxylation of carboxylic acids. Tyrosine and methionine residues are also affected.
crystallization papers

A preliminary X-ray characterization of the secreted 105 kDa glycoprotein β-mannosidase from the fungus T. reseei is reported.
d(ACGTAGCTACGT)2:[actinomycin D, (echinomycin)2] and d(ACGTAGCTACGT)2:[actinomycin D, (triostin A)2] have been crystallized into a monoclinic space group C2, with unit-cell dimensions a = 85.6, b = 72.8, c = 56.6 Å, β = 101.5° at 93 K and Z = 8. The Patterson maps calculated with 3 Å resolution data indicate that all complexes in the crystal are oriented along their helical axes in the [10 ] direction.
] direction.
Two different crystal forms of 1-L-myo-inositol-1-phosphate synthase (MIP synthase) from S. cerevisiae have been obtained. Crystals belonging to space group C2 (form I) diffract to 2.5 Å resolution and crystals belonging to space group P21 (form II) diffract to 2.9 Å resolution.

Crystals of pectate lyase A from E. chrysanthemi, a plant virulence factor, have been obtained and shown to diffract to 2.4 Å.

Native and iodinated T. molitor antifreeze proteins produced two different crystal forms. Crystals of iodinated TmAFPs diffracted to ∼1.4 Å resolution and belonged to the space group P61 (or P65) with unit-cell parameters a = 73.85, b = 73.85, c = 53.15 Å.

Monoclinic crystals of a thermostable DNA ligase from T. filiformis have been obtained. Native data have been collected to 3.0 Å using synchrotron X-rays.

Two orthorhombic crystal forms of the catalytic adenylate cyclase domain of GRESAG4.1 from T. brucei have been generated by the hanging-drop vapour-diffusion method.

The recombinant form of β-cinnamomin, a 10 kDa protein, was overproduced and crystallized. Crystals belonged to triclinic space group and diffracted to 1.45 Å

The crystallization and preliminary X-ray analysis of liganded (carboxy) haemoglobin from the South American freshwater fish known in Brazil as `pacu' is reported.
short communications

A rigid dimeric structure in the crystal shows some discrepancies from a monomeric NMR structure, which bring about the difference in the binding mode of FMN.
PDB reference: FNM-bp, 1flm

The structure ATP N indicates that the structural variations in class III plant peroxidases are restricted to one half of the molecule.
PDB reference: ATP N, 1qgj
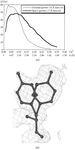
A space-grown crystal of H. lineatum collagenase has been recorded at 1.7 Å resolution. The newly refined crystal structure suggests a gate mechanism implicating tyrosine 99.
PDB reference: collagenase, 2hlc
books received
Free 

Free

Free

Free

Free









