

 journal menu
journal menu





issue contents
September 2003 issue

Cover illustration: Protein interactions of the phenylhydrazine adduct of the cysteine tryptophylquinone redox cofactor in quinohemoprotein amine dehydrogenase from Pseudomonas putida. C atoms are grey, N atoms are cyan, O atoms are red and S atoms are green (p. 1551).
research papers




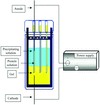
The influence of an internal electric field upon the protein structure of crystals grown inside capillary tubes is evaluated during the crystallization process by means of the gel-acupuncture method using lysozyme and thaumatin.


The crystal structure of PTR1 from L. tarentolae as a binary complex with NADPH at 2.8 Å resolution has been solved by molecular-replacement techniques using the recently reported L. major PTR1 structure as a search model.
PDB reference: L. tarentolae PTR1, 1p33, r1p33sf

A new crystal form of auracyanin B, a `blue' copper protein from the thermophilic green photosynthetic bacterium C. aurantiacus, has been refined at 1.9 Å resolution.
PDB reference: auracyanin B, 1ov8, r1ov8sf
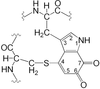
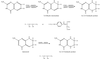
The binding site for phenylhydrazine in the quinohemoprotein amine dehydrogenase has been identified as the C6 position of the redox cofactor cysteine tryptophylquinone. This result firmly establishes C6 as the site of Schiff-base formation during catalytic turnover of this enzyme.
An estimator of weights for restrained macromolecular refinement is proposed which is based on an approximation of a marginal likelihood function of the weighting parameters and which uses all the X-ray data.
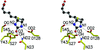
Mutation of residues in streptavidin capable of hydrogen bonding to biotin causes significant changes in the binding constants for this protein/ligand system. Structure analyses show few changes in the three-dimensional structures of biotin complexes with the mutated proteins.
PDB references: streptavidin mutant N23A, 1n4j, r1n4jsf; biotin complex of N23A, 1n43, r1n43sf; mutant N23E, 1n7y, r1n7ysf; mutant S27A, 1n9y, r1n9ysf; biotin complex of S27A, 1n9m, r1n9msf; mutant Y43A, 1nbx, r1nbxsf; 2-iminobiotin complex of Y43A, 1nc9, r1nc9sf; mutant Y43F, 1swu, r1swusf; biotin complex of Y43F, 1ndj, r1ndjsf

The 2.1 Å resolution structure of a king cobra PLA2 was determined by molecular replacement from a perfectly hemihedrally twinned crystal and compared with the structure of a homologous PLA2 from the same venom.
PDB reference: OH APLA2-II, 1m8t, r1m8tsf
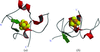
This paper describes the structure solution and the resultant model of R. fermentans HiPIP, a bacterial iron–sulfur protein with high reduction potential.
PDB reference: R. fermentans HiPIP, 1hlq, r1hlqsf
A crystal structure of guanidinoacetate methyltransferase in a monoclinic modification was determined in which three of four subunits have a catalytically unfavorable loop containing Tyr136. From analysis of the Gd ion-binding site, characteristic features of Gd-ion binding have been revealed and may be useful for preparing heavy-atom derivatives for phasing.

New diffraction data for phenol hydroxylase have been collected. Refinement to 1.7 Å resolution resulted in a final free R factor of 18.0%. The model was also corrected for 11 earlier sequencing errors.
PDB reference: phenol hydroxylase, 1pn0, r1pn0sf
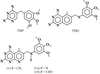
Structural data are reported for the first example of the potent antifolate inhibitor 2,4-diamino-5-methyl-6-[(3′,4′,5′-trimethoxy-N-methylanilino)methyl]pyrido[2,3-d]pyrimidine (1) in complex with human dihydrofolate reductase (hDHFR) and NADPH.

Because of the large number of molecules within a closely packed unit cell, this structure was solved by six-dimensional molecular replacement. Comparison with its counterpart from pea reveals highly conserved regions that are likely to be involved in intermolecular interactions.
PDB reference: T. thermophilus H-protein, 1onl, r1onlsf

A technique for automatically evaluating microbatch (sub-microlitre drop) protein crystallization trials is described.
structural genomics papers
A SARS-CoV whole-genome approach has been developed aimed at determining the crystal structure of all of its proteins or domains. Here, the cloning, E. coli expression, purification, and crystallization of the SARS-CoV Nsp9 protein, the first SARS-CoV protein to be crystallized, are reported.
crystallization papers

The native GroEL-GroES complex was isolated from T. thermophilus HB8 and crystallized. The triclinic crystal diffracted to 3.0 Å resolution.

An α-insect toxin from the scorpion B. martensii Karsch has been expressed, purified and crystallized.
Crystals suitable for structure determination of two enzymes in the biosynthesis pathway of anthracyclines in Streptomyces, aclacinomycin-10-methyl esterase and aclacinomycin-10-hydroxylase, have been grown.

Phosphoenolpyruvate carboxykinase (PCK) is a key enzyme involved in the regulation of gluconeogenesis. C. glutamicum PCK has been crystallized and X-ray diffraction data have been collected to 2.8 Å resolution.

ClpX from H. pylori has been overexpressed, purified and crystallized. The crystals diffracted to 2.6 Å resolution.

A probable ATP sulfurylase from T. thermophilus HB8 was overexpressed in E. coli and crystallized in the presence of adenosine-5′-phosphosulfate.
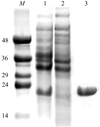
Crystals of nucleoside diphosphate kinase from rice have been obtained.

LysX protein, involved in the lysine-biosynthesis pathway in T. thermophilus, was cloned, expressed, purified and crystallized. Complete diffraction data sets were collected for the two crystal forms at 2.5 and 3.1 Å resolution. Structure determination is now in progress.

Methanogenic F420-dependent methylene-H4MPT dehydrogenase has been expressed, purified and crystallized. Crystals of selenomethionine-labelled enzyme diffracted to 1.5 Å resolution.
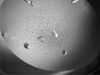
A two-domain construct of ZraR (HydG) response regulator from S. typhimurium has been crystallized and diffraction data have been recorded to 3.0 Å resolution.

Hyperthermophilic xylanase 10B from T. maritima MSB8 has been crystallized. A complete diffraction data set was collected up to 2.5 Å resolution.
The crystallization of and preliminary X-ray data for inorganic polyphosphate [poly(P)]/ATP-glucomannokinase from Arthrobacter sp. strain KM are presented.

The expression, purification, crystallization and preliminary X-ray diffraction studies of the monomeric pilin from P. aeruginosa strain K122-4 are reported.
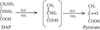
The PLP-dependent enzyme diaminopropionate ammonia lyase from E. coli has been crystallized in two forms. The monoclinic (P21) and the tetragonal (P41212 or P43212) forms diffracted X-rays to 2.5 and 3.0 Å resolution, respectively.

Native and SeMet derivatives of a type II cohesin from the cellulosomal system of A. cellulolyticus have been expressed, purified and crystallized in a trigonal form. Diffraction data were collected to 1.90 Å. In addition, orthorhombic crystals of the native protein were obtained that diffracted to 1.2 Å. Structures were solved using a combination of SIRAS and molecular replacement.

E. coli CcmG/DsbE has been crystallized using the vapour-diffusion technique and X-ray data have been collected to 1.9 Å resolution.
short communications

The crystal structure of UK114 has been determined, revealing a trimeric arrangement. It was confirmed by ultracentrifugation studies that UK114 is also a trimer in solution.
PDB reference: UK114, 1nq3, r1nq3sf







