

 journal menu
journal menu





issue contents
January 2005 issue
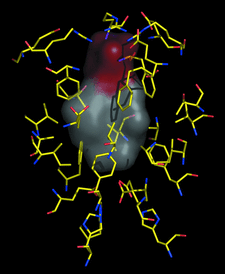
Cover illustration: Methyl-D-erythritol-2,4-cyclodiphosphate synthase (IspF) is involved in the biosynthesis of isoprenoid precursors. Results presented in this issue by Kemp et al. (p. p. 45) and also by Ni et al. in 2004 (Acta Cryst. D60, 1949-1957) indicate that the enzyme binds geranyl and farnesyldiphosphate at the core of a trimeric assembly.
editorial




Free 
research papers

A new algorithm to automatically fit a main-chain fragment to the electron density between two anchoring residues is presented. Examples of applications of the algorithm to the completion of initial protein models are included.


The crystal structure of an excitatory insect-specific toxin, BmK IT-AP, from the scorpion B. martensii Karsch determined at 2.6 Å resolution showed an extension and `wiggling' of the C-terminal segment (residues 59–72) with an additional one-and-a-half turn α-helix (α2) and a shifted disulfide bridge Cys38–Cys64 in comparison with the mammal-selective BmK α-toxins.
PDB reference: BmK IT-AP, 1t0z, r1t0zsf
High-resolution synchrotron X-ray powder diffraction data were used to determine binding modes for five N-acetylglucosamine oligosaccharides (NAGn, n = 2–6) on HEW lysozyme.

The influence of sampling a population of well defined protein conformations by small-angle scattering and subsequent data analysis is explored. The results demonstrate how the ensemble of well defined protein structures sampled influences the structural parameters and low-resolution models derived from the data.
Open access
access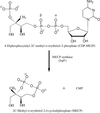
New crystal forms of 2C-methyl-D-erythritol-2,4-cyclodiphosphate synthase displayed electron density within an intersubunit cavity suggestive of an isoprenoid ligand. Mass spectrometry identified a mixture of ligands, the most abundant of which, geranyl diphosphate, was satisfactorily modelled in the binding sites. These ligands are produced downstream of this synthase in the non-mevalonate biosynthetic route to isoprenoids, suggesting the possibility of feedback regulation of this key metabolic pathway.

The effects of the silanized mica surface on protein crystallization have been investigated in the paper. It has been found that this silanized mica surface can ameliorate crystallization process considerably and improve the diffraction ability of crystals compared with the silanized glass cover slip.

Protein crystals can be clearly imaged and distinguished from non-protein crystals using the intrinsic ultraviolet fluorescence of these macromolecules. The method is fast and non-invasive and therefore well suited to the needs of high-throughput crystallography.

The random translocation of layers within a single crystal modulates observed intensities. A general forumla is proposed for solving this 50-year old problem.
Open access
access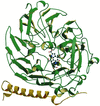
The crystal structure of methanol dehydrogenase (MDH) from Methylobacterium extorquens has been refined without stereochemical restraints at a resolution of 1.2 Å. The high-resolution data have defined the conformation of the tricyclic pyrroloquinoline quinone (PQQ) cofactor ring as entirely planar.
PDB reference: methanol dehydrogenase, 1w6s, r1w6ssf
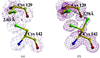
Cryogenic structures show systematic alterations from room-temperature structures. These cryoinduced structural changes include more extensive hydration, fewer disordered side chains, small coordinate shifts in the structural core and more extensive coordinate shifts at the protein surface.
PDB references: PAK pilin, 1x6z, r1x6zsf; 1x6x, r1x6xsf; 1x6q, r1x6qsf; 1x6p, r1x6psf; 1x6r, r1x6rsf; 1x6y, r1x6ysf
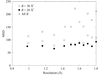
In this paper, the variation of the values of dihedral angles in proteins is divided into two categories by analyzing distributions in a database of structures determined at a resolution of 1.8 Å or better.
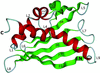
The structure of a pathogenesis-related PR-10 protein from a novel yellow lupin subclass confirms the general fold of this group of proteins, with a cavity within the protein core. The structure suggests that a segment of a long C-terminal α-helix characterized by low sequence conservation is involved in regulating the shape and size of the cavity, thus underlying the specificity of different PR-10 proteins towards their potential ligands.
PDB reference: pathogenesis-related PR-10 protein, 1xdf, r1xdfsf
short communications
Open access
access
Manganese phasing of a 225 kDa protein asymmetric unit is reported, demonstrating that, with modern synchrotron beamlines and software, very small anomalous signals from light metals, such as manganese, provide a practical tool for solving the structure of large proteins.
Open access
access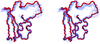
Thomas and Pastore describe a new algorithm that creates a single structure to represent ensembles produced by nuclear magnetic resonance spectroscopy or simulations of molecular dynamics.
international union of crystallography
Free








