journal menu
journal menu





issue contents
December 2006 issue
Cover illustration: Symmetry-related glycoside chains ordered because of tight mutual crystal packing (p. 1458).
research papers






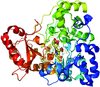
Structures of human protein tyrosine phosphatase β catalytic domain complexed with inhibitors were determined at high resolution. Engineering of protein variants suitable for the purposes of rapid structure-based drug discovery is discussed.

An automated high-throughput crystallization facility which is open to the general user community has been established at EMBL Hamburg. The facility provides more than 1000 initial crystallization conditions and the ability to design customized screens; it has the capacity to generate more than 100 96-well crystallization plates per day and to store and image up to 10 000 plates.
Open  access
access
access
The 1.3 Å resolution crystal structure of M. tuberculosis thioredoxin C is reported, demonstrating a novel packing of five C-terminal residues in the active-site groove.
PDB reference: thioredoxin C, 2i1u, r2i1usf
Open access
access
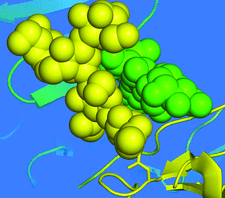
Crystals of native Cerezyme® (acid-β-glucosidase), the enzyme used in enzyme-replacement therapy in Gaucher disease, were obtained and the threee-dimensional structure solved. A number of sugar residues bound to three asparagines via N-glycosylation could be observed in the structure, and new conoformations of loops controlling access to the active site were detected.
PDB reference: acid-β-glucosidase, 2j25, r2j25sf
The crystal structure of E. coli UDP-N-acetylmuramoyl:L-alanine ligase (MurC) has been determined at 2.6 Å resolution by MAD.
PDB reference: MurC, 2f00, r2f00sf
S-SAD data from proteinase K at a wavelength of 0.98 Å with a Bijvoet ratio of ∼0.46% were successfully used for phasing of the macromolecular structure despite the presence of identifiable radiation-damage effects.
PDB reference: proteinase K, 2id8, r2id8sf

The high-resolution structure of the mouse p53 core domain, alone and bound to small-molecule compounds, provides a molecular scaffold for the structure-based design of p53-stabilization compounds for development as possible therapeutic agents.
Open access
access
The crystal structure of hydroxycinnamoyl-CoA hydratase-lyase (HCHL), the enzyme that catalyses the biotransformation of the coenzyme A thioester of ferulic acid [3-(4-hydroxy-3-methoxy-phenyl)prop-2-enoic acid] to vanillin (4-hydroxy-3-methoxy-benzaldehyde), has been solved and, in combination with a modelling study, a binding and discrimination mode for the substrate has been proposed.
PDB reference: HCHL, 2j5i, r2j5isf
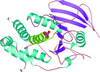
The crystal structure of the mouse UNC5H2 death domain at 2.1 Å resolution is reported. The UNC5H2 death domain is a dimer in the crystal and in solution.
PDB reference: UNC5H2 death domain, 1wmg, r1wmgsf
Open access
access
The heteromerization domain of the glutamyl-tRNA synthetase (GluRS) from yeast was crystallized and phase information was obtained from selenomethionine MAD data to 2.5 Å resolution. This structure and that of the interacting domain of Arc1p, a protein cofactor of GluRS, were refined to 1.9 Å resolution. Both domains adopt a GST-like fold, demonstrating a novel role for this fold as a protein–protein interaction domain.
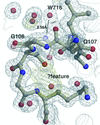
Comparison of three 1.1 Å resolution structures of Ffh reveals conserved patterns of surface-water interactions that accommodate both protein structural plasticity and the disruption of the hydrogen-bonding network by bound solutes.
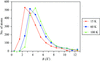
Increasing the temperature of X-ray data collection from 15 to 60 K results in an almost constant increase (∼1.7 Å2) in isotropic equivalents B of atomic displacement parameters for atoms with B values below 8 Å2.
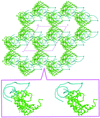
The crystal structure of ribosomal protein L1 in complex with a specific fragment of mRNA was solved at 2.1 Å resolution. Analysis of the crystal packing of L1–RNA complexes containing various 23S rRNA or mRNA fragments revealed a number of common features.
PDB reference: L1–mRNA, 2hw8, r2hw8sf

The structure of A. thaliana cell-wall invertase 1 was determined to be an N-terminal fivefold β-propeller domain followed by a C-terminal domain formed by two β-sheets.
PDB reference: cell-wall invertase 1, 2ac1, r2ac1sf
short communications
Open access
access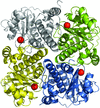
The crystal structure of NovR, a non-haem iron oxygenase from S. spheroides, was solved by molecular replacement with native X-ray data to 2.1 Å resolution using a template structure of relatively low sequence identity. An interpretable electron-density map was subsequently obtained from poor starting phases using a combination of fourfold averaging and very gradual phase extension.
addenda and errata
Open access
access