

 journal menu
journal menu





issue contents
July 2008 issue
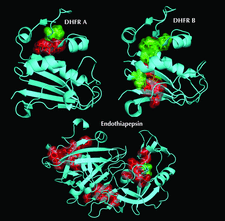
Cover illustration: Hydrophobic cores identified from HDX data and depth calculations of the neutron structures of DHFR and endothiapepsin (p. 764). The two DHFR monomers A and B represents the partially occluded and the closed forms, respectively. Proteins are shown in cyan and as cartoon representations. Residues which have backbone amides with depths ![[greater-than or equal to]](/logos/entities/ge_rmgif.gif) 3.8 Å are shown as semi-transparent surfaces and with side chains. Exchanged core residues are colored in green whereas non-exchanged core residues are shown in red.
3.8 Å are shown as semi-transparent surfaces and with side chains. Exchanged core residues are colored in green whereas non-exchanged core residues are shown in red.
research papers






The crystal structures of p38α MAP kinase complexed with two pyrrolotriazine-based inhibitors led to the elucidation of the high-affinity binding mode of these compounds.
Open  access
access
access
It is shown that the anisotropy of anomalous scattering (AAS) is a significant and ubiquitous effect in data sets collected at an absorption edge and that its exploitation can substantially enhance the phasing power of single- or multi-wavelength anomalous diffraction. The improvements in the phases are typically of the same order of magnitude as those obtained in a conventional approach by adding a second-wavelength data set to a SAD experiment.
Crystal structures of winged-bean basic agglutinin with disaccharides having glycosidic linkages that differ from those in the A and B blood-group substances reveal the structural basis for the higher specifity of the lectin for the blood-group antigens.

Using idealized known RNA secondary-structural fragments, it is demonstrated that it is possible to solve novel complex RNA structures without resort to heavy-atom phasing methods.
Crystal structures of M. tuberculosis folylpolyglutamate synthetase complexed with two nucleotides have been determined at 2.0 and 2.3 Å resolution, revealing an active-site loop movement and associated changes that influence substrate binding.
The high-resolution structure of HIV-1 subtype C protease contributes to the understanding of flap dynamics and drug resistance.
PDB reference: HIV-1 subtype C protease, 2r8n, r2r8nsf
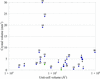
Backbone amide hydrogen/deuterium-exchange patterns can be directly observed from neutron crystal structures. For all the types of analysis performed, it is concluded that protection from exchange is only strongly correlated to secondary structure and the depth of the amide N atom.
The three-dimensional structure of human pancreatic carboxypeptidase A1 reveals the mode of binding of a polymer with drug-protecting effects and is used to apply optimal docking-area analysis to characterize binding sites for multiprotein complexes.
PDB reference: human carboxypeptidase A1, 2v77, r2v77sf
Separation of two individual lattices within an epitaxially twinned data set allowed the crystal structure of the V3-specific neutralizing antibody 447-52D in complex with a V3 peptide (UG1033) to be determined. The structure confirms that the neutralization breadth of Fab 447-52D is likely to be attributable to the extensive focus on main-chain hydrogen-bond interactions with the peptide that permit the recognition of a range of V3 sequences.
PDB reference: anti-HIV-1 Fab 447-52D–peptide complex, 3c2a, r3c2asf
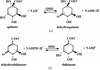
The crystal structure of an NAD+-dependent quinate dehydrogenase from C. glutamicum has been solved. Enzymatic assays show that this enzyme belongs to a new functional class of bacterial shikimate/quinate dehydrogenases.
PDB reference: NAD+-dependent quinate dehydrogenase, 2nlo, r2nlosf
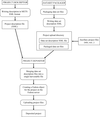
A suite of deposition tools have been created that allow X-ray diffraction images to be deposited in an open-access repository. The approach takes advantage of federated institutional repositories and aims to facilitate the archiving and sharing of raw X-ray diffraction images from the protein crystallography community.







