

 journal menu
journal menu





issue contents
April 2011 issue
From crystal to structure with CCP4
Proceedings of the CCP4 study weekend
Cover illustration: Human Thr160-phospho-CDK2-cyclin A complexed with a bis-anilino-pyrimidine inhibitor, PDB entry 2iw6. Drawn using CCP4mg (p. 386).




research papers
Open  access
access
access
An overview of the CCP4 software suite for macromolecular crystallography is given.
Open access
access
Protein surface engineering is increasingly used as a routine tool to enhance the crystallization propensity of proteins. Future possibilities include the use of multi-site protein variants, rational modulation of solubility and the development of strategies to tackle membrane proteins.
Open access
access
The Protein Information Management System (PiMS) is described together with a discussion of how its features make it well suited to laboratories of all sizes.
Open access
access
Thoughts about the decisions made in designing macromolecular X-ray crystallography experiments at synchrotron beamlines are presented.
Open access
access
A new graphical user interface to the MOSFLM program has been developed to simplify the processing of macromolecular diffraction data. The interface, iMOSFLM, allows data processing via a series of clearly defined tasks and provides visual feedback on the progress of each stage.
Open access
access
A summary of how to run the data-reduction programs in the CCP4 suite.
Open access
access
Typical topics and problems encountered during data processing of diffraction experiments are discussed and the tools provided in the autoPROC software are described.
Open access
access
The molecular-replacement model-improvement program Sculptor is described, with an analysis of the algorithms used.
Open access
access
The automated pipelines for molecular replacement MrBUMP and BALBES are reviewed, with an emphasis on understanding their output. Conclusions are drawn from their performance in extensive trials.
Open access
access
Some future challenges for the PDB and its guardians are discussed and current and future activities in structural bioinformatics at the Protein Data Bank in Europe (PDBe) are described.
Open access
access
Recent developments in the CRANK software suite for experimental phasing have led to many more structures being built automatically.

Open access
access
SAD data can be used in Phaser to solve novel structures, supplement molecular-replacement phase information or identify anomalous scatterers from a final refined model.
Open access
access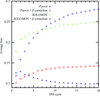
A cross-validation-based method for bias reduction in `classical' iterative density modification of experimental X-ray crystallography maps provides significantly more accurate phase-quality estimates and leads to improved automated model building.
Open access
access
The general principles behind the macromolecular crystal structure refinement program REFMAC5 are described.
Open access
access
The software suite Xsolve semi-exhaustively explores key parameters of the X-ray structure-determination process to compute multiple three-dimensional protein structures independently and in parallel from a set of diffraction images. An optimal consensus model for subsequent manual refinement is computed from these structures.
Open access
access
Methods for the analysis of the relationship between macromolecular complexes and interactions and their manifestation in crystal packing are described and discussed.
Open access
access
The CCP4 molecular-graphics program now uses the Qt framework to provide a modern look and feel. There are many new features including rendering for publication-quality images and sequence alignment.







