

 journal menu
journal menu





issue contents
September 2012 issue
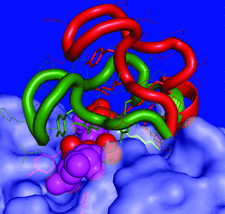
Cover illustration: Lid-loop motion in the active site of alginate lyase A1-III. The lid loop (residues 64-85) moves from an open to a closed form to make interactions with a bound substrate (spheres) and a catalytic Tyr residue (p. 1207).
research papers







The crystal structure of MoSub1, a Sub1/PC4 orthologue from rice blast fungus, has two novel features N-terminal and C-terminal to the DNA-binding domain. It has a similar dimer interface and DNA-binding region to PC4 and the protein binds single-stranded DNA tightly.
PDB reference: MoSub1, 4agh

Experimental errors in macromolecular crystallography are underestimated as they do not account for the contribution of crystal-to-crystal variations.
The crystal structures of the far-red fluorescent proteins eqFP650 and eqFP670 have been solved at 1.8 and 1.6 Å resolution, respectively. This permitted identification of the structural elements responsible for the bathochromic shift in both considered far-red fluorescent proteins.

Structural and functional studies show the importance of palmitic acid transport by the cytosolic protein ReP1-NCXSQ in regulation of the squid nerve Na+/Ca2+ exchanger NCXSQ1.
Dose-dependent atomic B factors are used to determine the average spatial distribution of radiation damage to crystalline thaumatin and urease.
The values of atomic shifts in unrestrained refinement can hint at alternative conformations.

The 2.4 Å crystal structure of the protein augmenter of liver regeneration (hsALR) containing a 14-residue histidine purification tag has been determined by Cd-SAD. The structure reveals a tetramer (two hsALR homodimers) bridged by a novel Cd2Cl4O6 cluster that produces a crystal packing environment that can better accommodate the N-terminal purification tag.
PDB reference: augmenter of liver regeneration, 3r7c
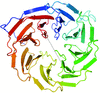
The BAM complex is comprised of an integral β-barrel outer membrane protein BamA and four accessory lipoproteins BamB, BamC, BamD and BamE. Here, the crystal structure of BamB is reported.
PDB reference: BamB, 3q54

Three crystal structures of a lipoprotein (Bmlp7) of unknown function, a member of the 30 kDa lipoprotein family from mulberry silkworm (B. mori L.) haemolymph, have been determined. The haemolymph-isolated protein was identified through successful sequence assignment according to electron-density maps at 1.33 Å resolution.

A method for experimental phasing by segmenting a large data set into sub-data sets for radiation-damage-induced phasing is presented.

The structure of a tetrameric sponge galectin suggests a basis for glutamate receptor potentiation.
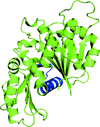
Structures of FtsZ from S. aureus in apo, GDP-bound and PC190723-complex forms are reported; FtsZ undergoes distinct conformational changes. These structural features are discussed and related to the crucial functions of FtsZ.

P99 cephalosporinase is a class C β-lactamase that is responsible in part for the widespread bacterial resistance to β-lactam antibiotics. Here, the X-ray crystal structure of the apo Asn152Gly mutant of P99 has been determined to 1.95 Å resolution.
PDB reference: P99 cephalosporinase, Asn152Gly mutant, 3s4x
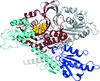
The structure of Giardia prolyl-tRNA synthetase cocrystallized with proline and ATP shows evidence for half-of-the-sites activity, leading to a corresponding mixture of reaction substrates and product (prolyl-AMP) in the two active sites of the dimer.
PDB reference: prolyl-tRNA synthetase, 3ial

A joint X-ray/neutron structure of D-xylose isomerase in complex with the inhibitor sorbitol was determined at room temperature at an acidic pH of 5.9. Protonation of the O5 O atom of the sugar was directly observed in the nuclear density maps. Under acidic conditions sorbitol gains a water-mediated interaction with the enzyme active site, which may explain the increased potency of the inhibitor at low pH.
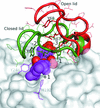
The structures of apo and holo mutants of alginate lyase A1-III revealed a large open/closed conformational change of lid loop. The closed lid loop is involved both in the binding of substrate and catalysis of the enzyme.
Determination of the orientation of the enterovirus 71 virions in the crystal required the calculation of a locked rotation function that included only icosahedral threefold and fivefold symmetry axes. Otherwise, misleading high rotation-function values were produced by accidental alignment of icosahedral and crystallographic twofold axes.
PDB reference: enterovirus 71, 4aed
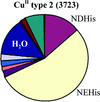
An updated picture of the ligand sets and copper–ligand atom bond lengths in proteins is presented.

The crystal structure of thymidylate synthase from E. faecalis was obtained as a native complex with 5-formyltetrahydrofolate (5-FTHF). The structure supports the role of 5-FTHF, which is naturally present in the bacterial cell, as a storage form of the folate metabolite and as an enzyme regulator.
PDB reference: thymidylate synthase, complex with 5-FTHF, 3uwl

The crystal structure of the 11.14 kDa orphan ORF 1382 from Archaeoglobus fulgidus (AF1382) has been determined by sulfur SAD phasing using data collected from a moderately diffracting crystal and 1.9 Å synchrotron X-rays.
short communications
Open  access
access
access
The surface of proteins can be charged with zinc ions and the anomalous signals from these zinc ions can be used for structure determination of proteins.







