

 journal menu
journal menu





issue contents
March 2013 issue
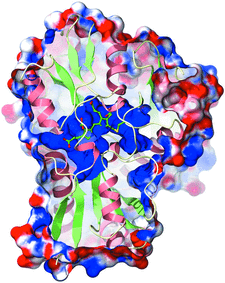
Cover illustration: The electrostatic surface of A. thaliana porphobilinogen deaminase (p. 471). The solvent-accessible surface of the enzyme is shown coloured according to the electrostatic potential. The surface has been clipped to show the large and highly electropositive (blue) binding site for the cofactor, which is formed predominantly of conserved arginine residues. The dipyrromethane and the cysteine residue to which it is attached (Cys254) are displayed in ball-and-stick representation.
research papers







The dimeric structure of Sfh3 (Sec14 family homologue 3 in yeast) is reported for the first time and differs from the Sec14 proteins reported to date, all of which are monomeric.
PDB reference: dimeric Sfh3, 4fmm
The structure of B. subtilis YwfH was determined at three conformational steps: the apo form, an apo-like conformation and the NADPH complex. The structures revealed synchronized conformational changes that facilitate cofactor binding and catalytic activity of the enzyme.
The crystal structure of the N-terminal domain of human thioredoxin-interacting protein (TXNIP), which belongs to a recently classified α-arrestin protein family, has been determined. This study provides the first structural information on any of the α-arrestins. Although TXNIP adopts a β-arrestin fold as predicted, it is structurally more similar to Vps26 proteins than to β-arrestins, while sharing below 15% pairwise sequence identity with either.
The plant Rab5 GTPase homologue ARA7 in complex with its guanine nucleotide-exchange factor VPS9a was crystallized in the presence of GDP and Ca2+, representing an initial intermediate of the exchange reaction.
PDB reference: ARA7-GDP-Ca2+–VPS9a, 4g01
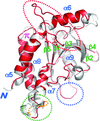
The mouse innate immune adaptor protein mSTING adopts a distinct conformation with two novel c-di-GMP binding modes.

The structure of virus-like particles of the small RNA phage PRR1 bound to an RNA segment corresponding to a stem-loop that includes the start codon for the replicase gene has been solved and the binding was compared with the related, and better investigated, phage MS2.A.
PDB reference: PRR1, complex with RNA segment, 4ang

The crystal structures of the N-terminal tandem RRM domains of HuR are reported in RNA-free and RNA-bound forms.


A six-axis robot arm equipped with a micro-gripper is used to harvest protein crystals from their crystallization drops. Once harvested, the sample is inserted into the X-ray beam by the robot for direct data collection.

The three-dimensional structure of a human IgA Fab is reported for the first time in three different crystal forms. Compared with a FabG with identical variable domains, the FabA displays a more rigid architecture which can explain the long-distance effects of the constant regions on antigen-binding affinity.
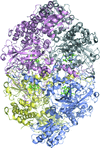
The catalytic and oxidative activities of the catalase from Scytalidium thermophilum are likely to be associated with the same haem active centre. The secondary oxidase activity may be a general feature of monofunctional catalases in the absence of hydrogen peroxide. This bifunctionality may provide some advantages for industry if the oxidase activity can be enhanced through engineering or directed evolution.

The binding of Rap1 to DNA induces potassium permanganate hypersensitivity that is driven by a specific Arg residue in Rap1 but is not associated with major DNA distortion.
PDB reference: Rap1–DNA complex, 4gfb
Open  access
access
access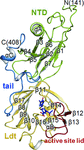
The crystal structure of M. tuberculosis L,D-transpeptidase (LdtMt2; Rv2518c) has been determined in both ligand-free and meropenem-bound forms. The detailed view of the interactions between meropenem and LdtMt2 will be useful in structure-guided discovery of new antituberculosis drugs.
Open access
access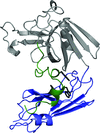
The crystal structures of two fragments of the L,D-transpeptidase from M. tuberculosis have been determined at 1.45 and 1.86 Å resolution. The extramembrane part of this enzyme consists of three domains: two domains related to the immunoglobulin fold and a catalytic domain belonging to the ErfK/YbiS/YhnG family at the C-terminus.
In this study, the crystal structure of AmyB from T. neapolitana has been determined at 2.4 Å resolution, revealing that the monomeric AmyB comprises domains A, B and C like other α-amylases, but with structural variations.
PDB reference: AmyB, 4gkl
Open access
access
The crystal structures of two 6-P-β-glucosidases from the GH1 family were determined in the apo form and in the presence of a 6′-P-salicin substrate, of the reaction product 6-P-β-glucose and of glucose corresponding to the aglycon molecule. The presence of natural ligands enabled the definition of the structural elements responsible for the recognition and hydrolysis of 6′-P-β-glucosides.
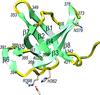
The crystal structure of the C-terminal domain of a putative U32 peptidase from G. thermoleovorans is reported; it is one of the most tightly packed protein structures reported to date.
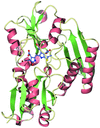
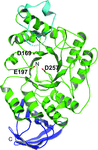
The first X-ray analysis of porphobilinogen deaminase from a plant reveals an extensive highly conserved loop region covering the active site that was completely disordered in previous structures. Structural comparisons suggest that the concerted movements of two enzyme domains may be linked to elongation of the substrate and that during the elongation cycle the bound dipyrromethane cofactor or polypyrrole intermediate moves to vacate one of the cofactor subsites such that an incoming pyrrole moiety can bind.
PDB reference: porphobilinogen deaminase, 4htg
short communications
Open access
access
A rational surface-engineering approach led to the crystal structure determination of ERK2–docking peptide complexes.
obituaries
Free








