

 journal menu
journal menu





issue contents
March 2014 issue

Cover illustration: 'Symmetrical merging' of the South rose window of the Gothic cathedral of Burgos in Northern Spain combined with the high symmetry axis view of the cylindrical enclosure (vault) formed by the major vault protein (MVP) from rat liver. Cathedral windows are magnificent examples of planar circular symmetry that are iconic representations of the cosmic view of the Middle Ages. Highly symmetrical atomic objects are frequent in biological structures, representing an atomic view of the biological microcosmos. Further details and discussion are given in the essay by C. Abad-Zapatero on p. 907. The images are from the author's personal collection and from T. Tsukihara and H. Tanaka, respectively.
research papers






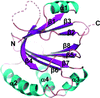
The structural and functional characterization of Isd-LmHde, an Isd-type haem-degrading enzyme in Listeria monocytogenes, is presented. The haem-degradation product of Isd-LmHde was verified to be biliverdin and the active residues were identified by structure-based mutagenesis and following function studies.
PDB reference: Isd-LmHde, 4kia
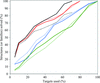
Using an extended set of protein features calculated separately for protein surface and interior, a new version of XtalPred based on a random forest classifier achieves a significant improvement in predicting the success of structure determination from the primary amino-acid sequence.
Open  access
access
access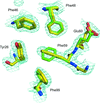
Two crystal forms of unligated FKBP12.6 exhibit multiple conformations in the active site and in the 80s loop, the primary site for known protein-recognition interactions. The previously unreported NMR backbone assignment of FKBP12.6 revealed extensive doubling of amide resonances, which reflects a slow conformational transition centered in the 80s loop.
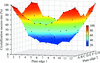
A novel and simple protein crystallization method called the cross-diffusion microbatch method is presented. This method can dramatically increase the number of crystallization conditions and is a potentially useful technique in practical protein crystallization screening.
Four high-resolution crystal structures of aIF2γ from Sulfolobus solfataricus are reported: aIF2γ-GDPCP (a nonhydrolyzable GTP analogue), aIF2γ-GDP-formate (in which a formate ion possibly mimics Pi), aIF2γ-GDP and nucleotide-free aIF2γ. The structures describe the different states of aIF2γ and demonstrate the conformational transitions that take place in the aIF2γ `life cycle'.
PDB references: aIF2γ–GDP, 4m0l; nucleotide-free aIF2γ, 4m2l; aIF2γ–GDP–formate, 4m4s; aIF2γ–GDPCP, 4m53

The structure of a peptide–peptoid analogue inhibitor was determined by X-ray crystallography. It is the first structure to reveal the structural basis of the complete proteolytic resistance towards serine proteases by inhibitors containing a peptoid residue at the P1 position.
PDB reference: bovine trypsin, complex with peptide–peptoid inhibitor, 4hgc

A low-activity mutant of a cold-adapted chitinase from M. marina has been crystallized and examined in an unliganded form and in complex with four and five N-acetyl-D-glucosamine units to reveal its substrate-binding mode. The oligomerization and conformational flexibility of this multi-domain protein in solution has also been examined.
An accumulation of severely non-isomorphous native data sets using the swelling-and-shrinking method or during search for the isomorphous replacement solutions can be beneficial for phase improvement and perhaps for structure refinement.
PDB reference: G-segment invertase–DNA complex, 4m6f
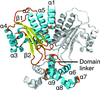
Structural insights into the molecular mechanism of Escherichia coli SdiA, a quorum-sensing receptor
Crystal structures of the full-length E. coli quorum-sensing receptor SdiA reveal that it forms a symmetrical dimer and exhibits broad ligand-binding selectivity. The ligand binding increases the stability of SdiA but does not modulate its DNA-binding ability. The DNA-binding activity of SdiA can be regulated through the formation of an intersubunit disulfide bond.
Open access
access
Catalytic antibody variants with κ and λ light-chain constant domains show differences in their crystal structures which lead to subtle changes in catalytic efficiency and thermodynamic parameters as well as in their affinity for peptide substrates.
PDB reference: A17λ, 3zl4

Crystal structures of the Y138F obelin mutant before and after bioluminescence have been determined at 1.72 and 1.30 Å resolution, respectively. Their spatial structures support the catalytic function of a water molecule found in an internal cavity in the light-emission reaction of Ca2+-regulated photoproteins.
Open access
access
Ensemble-refinement analysis of native and mutant factor D (FD) crystal structures indicates a dynamical transition in FD from a self-inhibited inactive conformation to a substrate-bound active conformation that is reminiscent of the allostery in thrombin. Comparison with previously observed dynamics in thrombin using NMR data supports the crystallographic ensembles.
Open access
access
The enzyme porphobilinogen deaminase (PBGD; hydroxymethylbilane synthase; EC 2.5.1.61) catalyses a key early step in the biosynthesis of tetrapyrroles in which four molecules of the monopyrrole porphobilinogen are condensed to form a linear tetrapyrrole. Two near-atomic resolution structures of PBGD from B. megaterium are reported that demonstrate the time-dependent accumulation of partially oxidized forms of the cofactor, including one that possesses a tetrahedral C atom in the terminal pyrrole ring.
Open access
access
The structure of a spore photoproduct lesion in duplex DNA is described.

The crystal structure of the class C extended-spectrum cepahalosporinase ADC-1 has been solved to 1.2 Å resolution.
PDB reference: ADC-1 β-lactamase, 4net
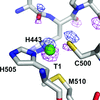
In the present study, crystal structures of multicopper oxidases were determined and density for an O atom was found inside the trinuclear copper centre.

The crystal structure of the dimer form of CTNNBL1 was determined, and the CDC5L binding site was also identified. Based on the structural and the biochemical experiments of CTNNBL1, an improved molecular architecture of the Prp19–CDC5L complex was presented.

A metal-independent glutaminyl cyclase from the black-legged tick I. scapularis is reported. The metal-independent inhibition of glutaminyl cyclases by imidazole-derived inhibitors is described for the first time.

Structural insights into L. major farnesyl diphosphate synthase, a key enzyme in the mevalonate pathway, are described.
Open access
access
In order to clarify the structural basis of the halophilic characteristics of an alkaline phosphatase derived from the moderate halophile Halomonas sp. 593 (HaAP), the tertiary structure of HaAP was determined to 2.1 Å resolution by X-ray crystallography. The structural properties of surface negative charge and core hydrophobicity were shown to be intermediate between those characteristic of halophiles and non-halophiles, and may explain the unique functional adaptation to a wide range of salt concentrations.
PDB reference: HaAP, 3wbh

The structure of Rv2372c from M. tuberculosis reveals it to be an RsmE orthologue that belongs to a distinct class in the SPOUT superfamily and can complement RsmE-deleted E. coli cells.
PDB reference: Rv2372c, 4l69

This work reports the first sub-angstrom resolution structure of S. erythraeus trypsin. The detailed model of a prototypical serine protease at a catalytically relevant pH with an unoccupied active site is presented and is compared with other high-resolution serine protease structures.
PDB reference: Streptomyces erythraeus trypsin, 4m7g

The structure of SAICAR synthetase from S. pneumoniae had been solved in complex with ADP, Asp, Mg2+ and AIR. The newly defined binding site for Asp provides information about the mechanism.
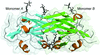
The crystal structure of Human cytomegalovirus immune modulator UL141 was solved at 3.25 Å resolution. Here, a detailed analysis of its intimate dimerization interface and the biophysical properties of its receptor (TRAIL-R2 and CD155) binding interactions are presented.
PDB reference: UL141, 4jm0
Open access
access
Crystal structures of two truncated variants of the transcription factor PpsR from R. sphaeroides are presented that enabled the phasing of a triple PAS domain construct. Together, these structures reveal the importance of α-helical PAS extensions for multi-PAS domain-mediated protein oligomerization and function.
Open access
access
Substitutive mutations that convert a tetrameric β-glucosidase into a dimeric state lead to improvement of its crystal quality.
PDB reference: BGLPf-M3, 3wdp

The polyfluorinated compound 2,2′,3,3′,5,5′,6,6′-octafluoro-4,4′-biphenyldiol (JF0064) is a potent inhibitor (IC50 ≤ 1 µM) of both aldose reductase and AKR1B10. X-ray structures of ternary complexes of both enzymes with JF0064 allow it to be inferred that this lead compound binds to the enzyme active site through an acidic phenol group and allow the extraction of important hints for structure-based drug design.
short communications
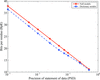
Information theory is used to estimate the precision of reported protein coordinates in the PDB.
essays
Free

The magnificent rose windows of the Gothic cathedrals provide artistic illustrations of many of the planar point groups of symmetry. It is suggested that the presence of certain symmetry elements and their selection by the artists could have been motivated to convey specific meaning to the resulting compositions.







