

 journal menu
journal menu





issue contents
April 2014 issue
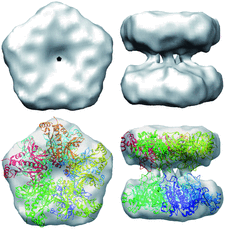
Cover illustration: Electron cryomicroscopy reconstruction of the plastid-located glutamine synthetase GSII-2a from Medicago truncatula and the fitting of the crystal structure of the cytoplasmic GSII-1a from M. truncatula, with each monomer coloured differently (p. 981).
research papers






The putative gene smu.1420 from S. mutans is demonstrated to encode the MdaB protein with NADPH-specific quinone oxidoreductase activity. Crystal structures of Smu.1420 with NADP+ and menadione reveal the substrate-selection and catalytic mechanisms, and shed light on future drug development against S. mutans.
IPM isomerase and homoaconitase belong to the aconitase family of enzymes and are composed of large and small subunits. The large subunits of the two enzymes adopt different active-site states before Fe–S cluster binding.
Open  access
access
access
The first crystal structure of an insect chitinase that is indispensable to moulting is revealed.

The 2.7 Å resolution crystal structure of the CcbJ methyltransferase from S. caelestis, which is part of the biosynthetic pathway of the lincosamide antibiotic celesticetin, is reported along with its complex with S-adenosyl-L-homocysteine at 2.9 Å resoution. Mutational and docking studies based on these structures are used to propose a plausible mechanism for its activity.
Open access
access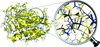
Paramagnetic NMR data (pseudocontact shifts and self-orientation residual dipolar couplings) and diamagnetic residual dipolar couplings can now be used in the program REFMAC5 from CCP4 as structural restraints together with X-ray crystallographic data. These NMR restraints can reveal differences between solid state and solution conformations of molecules or, in their absence, can be used together with X-ray crystallographic data for structural refinement.

The structure of Δ1-pyrroline-5-carboxylic dehydrogenase (PruA), involved in proline utilization in M. tuberculosis, has been determined in apo and NAD+-bound forms. Opportunities for inhibitor development are identified and the proline-utilization pathway has been characterized with a novel use of NMR.
Open access
access
The experimental models of dicotyledonous cytoplasmic and plastid-located glutamine synthetases unveil a conserved eukaryotic-type decameric architecture, with subtle structural differences in M. truncatula isoenzymes that account for their distinct herbicide resistance.
PDB reference: glutamine synthetase, 4is4

The crystal structure of 3-dehydro-3-deoxy-D-gluconate 5-dehydrogenase from T. thermophilus HB8 has been determined in the apo form, as well as in complexes with the cofactor and with citrate, by X-ray diffraction methods. The citrate molecule and the cofactor have been identified deep in the cleft formed between the interface of two adjacent subunits. These structures provide insights into the substrate recognition and catalytic machinery of 3-dehydro-3-deoxy-D-gluconate 5-dehydrogenase.
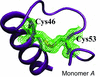
The crystal structure of wild-type human neuroglobin reveals two distinct conformations of the CD loop that contains an internal disulfide bridge. The increased affinity for oxygen upon disulfide-bond formation may be explained by the flexibility of the CD loop and the remodelling of the internal cavities.
PDB reference: neuroglobin, 4mpm
Open access
access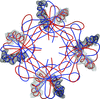
Crystal lattice disorders are a phenomenon which may hamper the determination of macromolecular crystal structures. Using the case of the crystal structure of stefin B, identification of rotational order–disorder and structure determination are described.
PDB reference: stefin B, 4n6v
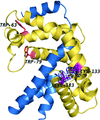
A disulfide bond in the −35 promoter-binding domain of σK regulates its interaction with the anti-σ factor RskA.
PDB reference: SigK–RskA complex, 4nqw
Open access
access
The side-chain torsion angles of isoleucines in X-ray protein structures are a function of resolution, secondary structure and refinement software. Detailing the standard torsion angles used in refinement software can improve protein structure refinement.
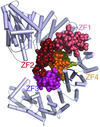
The X-ray structure of the complex of the nuclear transporter importin β with the zinc-finger-type transcription regulator Snail1 is reported.
PDB reference: Snail1–importin β complex, 3w5k
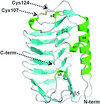
The high-resolution crystal structure of FfIBP was determined and the structure showed an intramolecular disulfide bond in the capping head loop region and a T-A/G-X-T/N ice-binding motif. The rigid capping head loop region and greater surface area of the ice-binding site are important for the antifreeze activity of FfIBP.
The crystal structures of Y. pestis RipA mutants were determined to provide insights into the CoA transferase reaction pathway.

Crystallographic characterization of the (R)-selective amine transaminase from Aspergillus fumigatus
The X-ray crystal structure of the (R)-selective amine transaminase from A. fumigatus at 1.27 Å resolution was determined via S-SAD phasing at 1.84 Å resolution.
PDB reference: (R)-selective amine transaminase, 4chi
The first crystal structure of the VgrG3CCD-TsiV3 complex has been determined. The detailed biochemical, biophysical and functional studies delineated the structural basis for self-protection in Vibrio cholerae.
PDB reference: VgrG3CCD–TsiV3 complex, 4noo
Open access
access
The solvent-picking procedure in phenix.refine has been extended and combined with Phaser anomalous substructure completion and analysis of coordination geometry to identify and place elemental ions.
A combination of powder diffraction and macromolecular crystallography demonstrated the presence of at least three different crystal forms of B. lentus subtilisin in a suspension from a large-scale industrial production. The application of powder diffraction is a powerful tool for quality control in enzyme production.

The 3 Å resolution structure of the type IV secretion system protein TraK from Gram-positive conjugative plasmid pIP501 is reported.
PDB reference: TraK, 4hic
Open access
access
Chemical bonding at the active site of lysozyme is analyzed on the basis of a multipole model employing transferable multipole parameters from a database. Large B factors at low temperatures reflect frozen-in disorder, but therefore prevent a meaningful free refinement of multipole parameters.
Crystal structures of L. pneumophila NTPDase1 in complex with three polyoxometallate clusters suggest different inhibitory mechanisms.
X-ray crystallography shows that 3′-azidothymidine, a reversible inhibitor of E. coli thymidine phosphorylase, is bound in the active site of the enzyme in an orientation different from that of the thymidine substrate and nucleoside analogues in known complexes of pyrimidine nucleoside phosphorylases. The possible role of the 3′-azido group as a trigger of reorientation of the ligand is discussed.
PDB reference: thymidine phosphorylase, complex with AZT, 4lhm
Open access
access
Threonine 57 is identified as the key residue required for the post-translational activation of E. coli aspartate decarboxylase. The crystal structure of the site-directed mutant T57V is reported.
PDB reference: aspartate decarboxylase, T57V mutant, 4azd
addenda and errata
Free

A correction is made to the article by Singh et al. [(2014). Acta Cryst. D70, 752–759].
Free

A number of corrections are made to the article by Bibby et al. (2012). Acta Cryst. D68, 1622–1631.
book reviews
Open access
access







