

 journal menu
journal menu





issue contents
September 2014 issue
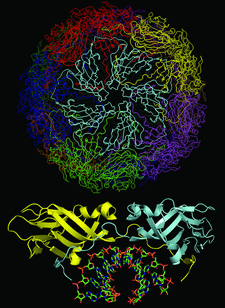
Cover illustration: Satellite tobacco mosaic virus refined to 1.4 Å (p. 2316). The full capsid structure is illustrated together with a dimer and its associated RNA double helix.
feature articles




Open  access
access
access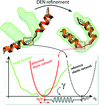
An overview of applications of the deformable elastic network (DEN) refinement method is presented together with recommendations for its optimal usage.
research papers


Comparison of mutant and wild-type hammerhead ribozyme structures reveals active-site structural perturbations consistent with either a general base catalytic mechanism, as previously assumed to be operative in the hammerhead ribozyme, or a specific base catalytic mechanism, which may possess additional explanatory power.
PDB reference: schistosomal hammerhead ribozyme, G12A mutant, 3zd4
The 2.40 Å resolution crystal structure of MSMEG_5817 revealed structural homology to sterol carrier proteins, suggesting a lipid-transport role in the survival of mycobacteria within host macrophages.
PDB reference: MSMEG_5817, 4nss
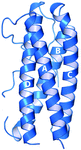
Interleukin-11 is a multifunctional member of the interleukin-6 family of cytokines. The crystal structure of human interleukin-11 is reported, providing important structural details of the receptor binding regions and revealing structural differences form the archetypal family member, interlekin-6.
PDB reference: human interleukin-11, 4mhl
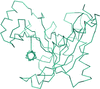
A 2.3 Å resolution crystal structure of human EndoV was determined and the structural basis of its unusual substrate specificities was explored.
PDB reference: human endonuclease V, 4nsp
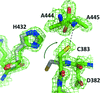
RipA is a key cysteine protease of Mycobacterium tuberculosis as it is responsible for bacterial daughter-cell separation. By combining crystallographic and mutational studies with functional assays and molecular modelling, it is shown that the catalytic activity of the enzyme relies on a Cys–His–Glu triad. The impact of the mutation of each residue of the triad on the structure and function of RipA is also analysed.
Open access
access
Mushroom tyrosinase isoform abPPO4 (Agaricus bisporus polyphenol oxidase 4) was crystallized by means of an Anderson-type polyoxometalate. The enzyme crystallized as a crystallographic heterodimer containing the zymogen (L-TYR; 64 kDa), the 21 kDa smaller activated form (A-TYR) and the polyoxometalate (POM) within one single crystal in a 1:1:1 ratio.
PDB reference: abPPO4 mushroom tyrosinase, 4oua

The crystal structure of Satellite tobacco mosaic virus (STMV) at 1.4 Å resolution represents the highest resolution and most precise native virus structure, utilizing more than twice as many reflections in refinement as were used for the previous STMV model. New details regarding the multiple ion arrangement on fivefold axes, the disposition of the `free' nucleotide, alternate conformations of amino-acid side chains and the distortion of the capsid from strict icosahedral symmetry have emerged.
Open access
access
A novel direct phase-selection method to select optimized phases from the ambiguous phases of a subset of reflections to replace the corresponding initial SAD phases has been developed. With the improved phases, the completeness of built residues of protein molecules is enhanced for efficient structure determination.
Open access
access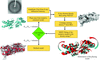
A new real-space refinement method for low-resolution X-ray crystallography is presented. The method is based on the molecular dynamics flexible fitting protocol targeted at addressing large-scale deformations of the search model to achieve refinement with minimal manual intervention. An explanation of the method is provided, augmented by results from the refinement of both synthetic and experimental low-resolution data, including an independent electrophysiological verification of the xMDFF-refined crystal structure of a voltage-sensor protein.
Open access
access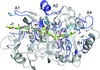
Cellobiohydrolase Cel7A from H. grisea var. thermoidea showed a 10°C higher Tm and a 75% higher yield than H. jecorina Cel7A in a performance assay at 65°C. The crystal structure at 1.8 Å resolution indicates higher flexibility in tunnel-defining loops and reveals a new loop conformation near the active centre.
PDB reference: Cel7A, 4csi

The recorded pH of protein crystallization screens is often inaccurate. Here, a method has been developed for the high-throughput measurement of pH for these screens using an acid–base indicator and a spectrophotometer.
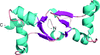
The TraN structure shows an internal dimer fold with helix–turn–helix motifs at both ends. The protein is a double-stranded DNA-binding protein that binds upstream of the pIP501 oriT, presumably acting as a type IV secretion-system regulator involved in early steps of conjugative transfer.

Open access
access
Commercially available adhesives have been evaluated for crystal mounting when undertaking complex macromolecular crystallography experiments. Here, their use as tools for advanced sample mounting and cryoprotection is assessed and their suitability for room-temperature data-collection and humidity-controlled studies is investigated.
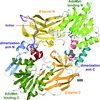
The crystal structure of full-length PRMT7 from M. musculus refined at 1.7 Å resolution is reported and provides structural clues for the monomethylation of arginine.
PDB reference: PRMT7, 4c4a
Open access
access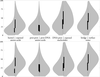
Distributions of scaled B factors from 704 protein–DNA complexes reflect primarily the neighbourhood of amino-acid and nucleotide residues: their flexibility grows from the protein core to protein–protein and protein–DNA interfaces, to solvent-exposed residues. Some of the findings clearly observed at higher resolution structures can no longer be observed for structures at low resolution indicating problems in refinement protocols.
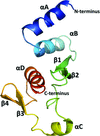
The structure of the C-terminal domain of the Zaire ebolavirus nucleoprotein (NP), encompassing residues 641–739, was determined in two distinct crystal forms. The domain has a novel fold that is distantly related to the β-grasp fold.

Pseudo-symmetry, which is common in macromolecular crystals, may result in incorrect space-group assignment that remains unnoticed until the advanced stages of refinement. The program Zanuda has been developed to automate space-group assignment and validation.
Open access
access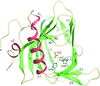
The first X-ray structure of a 2,4′-dihydroxyacetophenone dioxygenase from Alcaligenes sp. 4HAP at a resolution of 2.2 Å is reported. This structure establishes that the enzyme adopts the cupin-fold, forming compact dimers with a pronounced hydrophobic interface between the monomers. Each monomer possesses a catalytic ferrous iron that is coordinated by three histidines (76, 78 and 114) and an additional ligand which has been putatively assigned as a carbonate, although formate and acetate are possibilities.
PDB reference: 2,4′-dihydroxyacetophenone dioxygenase, 4p9g

The crystal structure of Est-Y29, a metagenomic homologue of the penicillin-binding protein/β-lactamase family, was determined at 1.70 Å resolution. The biochemical properties of Est-Y29 were also investigated for potential biotechnological applications.

The crystal structure of Rot shows a unique pattern of dimerization that differs from that of other SarA homologues.
PDB reference: Rot, 4q77
Open access
access
The crystal structure of PvuRts1I was determined and a 5-hydroxymethylcytosine-binding pocket was identified in the SRA-like domain. Enzyme variants were engineered to assist in hydroxymethylome mapping based on the crystal structure of PvuRts1I.
PDB reference: PvuRts1I, 4oky
Open access
access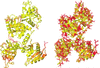
The Procrustes Structural Matching Alignment and Restraints Tool (ProSMART) has been developed to allow local comparative structural analyses independent of the global conformations and sequence homology of the compared macromolecules. This allows quick and intuitive visualization of the conservation of backbone and side-chain conformations, providing complementary information to existing methods.







