
 journal menu
journal menu










research papers
 | STRUCTURAL BIOLOGY |
 access
accessSmall-angle neutron scattering reveals the assembly mode and oligomeric architecture of TET, a large, dodecameric aminopeptidase

aUniversité Grenoble Alpes, IBS, 38044 Grenoble, France, bCNRS, IBS, 38044 Grenoble, France, cCEA, IBS, 38044 Grenoble, France, dLife Sciences Group, Institut Laue–Langevin, 38042 Grenoble CEDEX 9, France, and eLarge Scale Structures Group, Institut Laue–Langevin, 38042 Grenoble CEDEX 9, France
*Correspondence e-mail: [email protected]
The specific self-association of proteins into oligomeric complexes is a common phenomenon in biological systems to optimize and regulate their function. However, de novo of these important complexes is often very challenging for atomic-resolution techniques. Furthermore, in the case of homo-oligomeric complexes, or complexes with very similar building blocks, the respective positions of subunits and their assembly pathways are difficult to determine using many structural biology techniques. Here, an elegant and powerful approach based on small-angle neutron scattering is applied, in combination with deuterium labelling and contrast variation, to elucidate the oligomeric organization of the and the assembly pathways of 468 kDa, hetero-oligomeric and symmetric Pyrococcus horikoshii TET2–TET3 aminopeptidase complexes. The results reveal that the topology of the PhTET2 and PhTET3 dimeric building blocks within the complexes is not casual but rather suggests that their quaternary arrangement optimizes the catalytic efficiency towards peptide substrates. This approach bears important potential for the determination of quaternary structures and assembly pathways of large oligomeric and symmetric complexes in biological systems.
Keywords: TET; small-angle neutron scattering.
1. Introduction
The specific self-association of proteins to form oligomeric machines is a common phenomenon in biological systems. Homo-oligomerization in a closed symmetry is particularly prevalent in enzymes and high-order oligomers comprising more than six subunits and represents a significant part of the proteomes (Matthews & Sunde, 2012![[Matthews, J. & Sunde, M. (2012). Protein Dimerisation and Oligomerisation in Biology, edited by J. Matthews, pp. 1-18. New York: Springer.]](../../../../../../logos/arrows/d_arr.gif "Matthews, J. & Sunde, M. (2012). Protein Dimerisation and Oligomerisation in Biology, edited by J. Matthews, pp. 1-18. New York: Springer.") ). There are numerous reasons why enzymes self-assemble into large edifices: while representing a way to increase the stability and solubility of the system, the main advantage afforded by complex quaternary structures is to improve enzymatic function. Subunit interactions often allow the formation of channels to increase the specific affinity towards the substrate(s). Oligomerization also induces cooperativity between monomers to build up active sites or to share them at interfaces (Marianayagam et al., 2004) and, in multi-subunit enzymatic complexes, the quaternary structure enhances functional cooperativity (Griffin & Gerrard, 2012). Monomers assembled into large, hollow edifices can create compartments for the confinement of biochemical activities. In the case of energy-dependent chaperonins or complexes of ATPases associated with various cellular activities (AAA-ATPases), this strategy is exploited to unfold, refold or disassemble macromolecular edifices in the interior of a nano-compartment, thus avoiding aggregation in the crowded cytosolic environment (Snider & Houry, 2008). In the case of large intracellular peptidases such as proteasomes, the self-compartmentalization allows the confinement of the active sites inside the final particle and, in this way, uncontrolled proteolytic activity is avoided in the cytosol (Sauer & Baker, 2011). Finally, another advantage of oligomerization is to generate docking platforms for regulatory proteins or complexes. For all of these reasons, large enzymatic machines must adopt a well defined quaternary structure to carry out their function, and one way to regulate their activity within the cell is by controlling their oligomerization state.
). There are numerous reasons why enzymes self-assemble into large edifices: while representing a way to increase the stability and solubility of the system, the main advantage afforded by complex quaternary structures is to improve enzymatic function. Subunit interactions often allow the formation of channels to increase the specific affinity towards the substrate(s). Oligomerization also induces cooperativity between monomers to build up active sites or to share them at interfaces (Marianayagam et al., 2004) and, in multi-subunit enzymatic complexes, the quaternary structure enhances functional cooperativity (Griffin & Gerrard, 2012). Monomers assembled into large, hollow edifices can create compartments for the confinement of biochemical activities. In the case of energy-dependent chaperonins or complexes of ATPases associated with various cellular activities (AAA-ATPases), this strategy is exploited to unfold, refold or disassemble macromolecular edifices in the interior of a nano-compartment, thus avoiding aggregation in the crowded cytosolic environment (Snider & Houry, 2008). In the case of large intracellular peptidases such as proteasomes, the self-compartmentalization allows the confinement of the active sites inside the final particle and, in this way, uncontrolled proteolytic activity is avoided in the cytosol (Sauer & Baker, 2011). Finally, another advantage of oligomerization is to generate docking platforms for regulatory proteins or complexes. For all of these reasons, large enzymatic machines must adopt a well defined quaternary structure to carry out their function, and one way to regulate their activity within the cell is by controlling their oligomerization state.
Surprisingly, only a few studies have addressed the fundamental questions of the assembly pathways of large homo-oligomers. Progress has been made in recent years, indicating that dimeric precursors are often involved (Marianayagam et al., 2004). However, the structural details and control mechanisms of the assembly processes are still largely unknown: are they orchestrated, stepwise processes with well defined oligomeric intermediates? Does assembly follow a limited number of pre-defined pathways or is it of a random nature? How are the intermediate oligomers positioned in the final complex? Concomitantly, the study of the assembly processes of homomeric, symmetric complexes poses two practical challenges: (i) the isolation, stabilization and biochemical/biophysical characterization of the building blocks and intermediate states and (ii) the determination of their respective positions and arrangement within the final edifice. Here, we apply a powerful and elegant approach based on the combination of small-angle neutron scattering (SANS), deuterium labelling and contrast variation to elucidate the oligomeric organization of the quaternary structures and the assembly modes of symmetric, heterododecameric, 468 kDa TET aminopeptidase complexes.
TET complexes are bi-metallic aminopeptidases that act as peptide-destruction machines and are widespread in the three domains of life. They belong to the M42 and M18 families in clan MH according to the MEROPS classification system (Rawlings et al., 2014). The typical TET tetrahedral quaternary structure was initially described in archaea (Franzetti et al., 2002; Russo & Baumann, 2004; Borissenko & Groll, 2005), but has also been found in bacteria (Kim et al., 2010) and in eukarya (Chen et al., 2012; Chaikuad et al., 2012). Unlike most homomers, which adopt cyclic or dihedral symmetries, TET peptidases display unusually sophisticated quaternary structures, with 12 monomers being arranged in a hollow, tetrahedral edifice (Franzetti et al., 2002). The crystallographic structures and the enzymatic properties of three different complexes (PhTET1, PhTET2 and PhTET3) from the hyperthermophilic archaeon Pyrococcus horikoshii have been determined (Durá et al., 2005, 2009; Schoehn et al., 2006). They form self-compartmentalized assemblies with a common internal organization made up of a network of four access channels extended by four vast catalytic chambers, each containing three active sites close to the tetrahedron apices (Durá et al., 2005). Each TET subunit is formed by a proteolytic and a PDZ-like domain that mediate both the dimeric interface and the dimer–dimer interface that build the whole tetrahedral particle. We recently demonstrated that the activity of TET aminopeptidase towards long polypeptides is coupled with its assembly process (Appolaire et al., 2013; Rosenbaum et al., 2011): the co-occurrence in vivo of stable TET dimeric precursors and assembled dodecamers suggests that, at least in archaea, the TET oligopeptidase activity is regulated by control of its state. PhTET homododecamers are very stable, even at high temperatures (90°C), a fact attributed to the properties of the dimer interfaces (Appolaire et al., 2013). In vivo studies, combined with small-angle X-ray scattering (SAXS), showed that dimers are the elementary building blocks and, based on (EM) and analytical ultracentrifugation (AUC) data, a working model with hexameric intermediates assembling into the final dodecameric edifices has been suggested (Appolaire et al., 2013).
The three M42 protein homomers comprised in the genome of P. horikoshii display complementary substrate specificities: PhTET1 is a glutamyl-aminopeptidase that is active towards acidic residues, PhTET2 is a leucyl-aminopeptidase that is active towards neutral residues, and PhTET3 is a lysyl-aminopeptidase that is mainly active towards basic residues. The three forms therefore represent an integrated cellular peptide-destruction system (Durá et al., 2009). PhTET2 and PhTET3 display a high sequence identity, are robust thermozymes and their quaternary structures resist harsh physicochemical treatments (Rosenbaum et al., 2012). Their assembly process is metal-dependent and, in vitro, addition of a metal chelator agent combined with high-pH conditions is necessary to disassemble the complex into its stable dimeric precursors. These can spontaneously reform the tetrahedral edifice upon dialysis against a physiological assembly buffer containing cobalt (Rosenbaum et al., 2011).
Here, we show that when mixed together in the assembly buffer PhTET2 and PhTET3 dimers can self-organize into 468 kDa heterododecameric complexes that possess the same quaternary structure as homododecameric TETs. Using a combination of SANS and contrast variation (variable H2O/D2O ratio in the solvent; Jacrot, 1976) on heterododecameric complexes of deuterated PhTET2 (`dPhTET2') and hydrogenated PhTET3 (`hPhTET3') allowed us to elucidate their respective quaternary architectures within the assembled hetero-complex. Our results demonstrate that TET complexes are built following a very limited number of specific and well defined pathways. Intriguingly, the resulting geometric arrangement of the two different building blocks within the heterododecameric complex is not casual but rather suggests a mechanism to optimize its catalytic properties towards peptide substrates. Our approach represents an elegant and attractive method to address structural questions in other challenging oligomeric systems composed of symmetric (or very similar) building blocks such as the proteasome. Finally, a better structural insight into the assembly mechanisms of large complexes is essential for preparing studies of the underlying regulatory mechanisms, which have not been intensively explored to date but might represent attractive targets for biomedical approaches and drug design.
2. Material and methods
2.1. Expression and purification of hydrogenated PhTET3 and deuterated PhTET2
Total protein extracts of Escherichia coli cells expressing the various recombinant hydrogenated PhTET3 proteins (hPhTET3) were purified as described by Durá et al. (2009). For SANS experiments, random-fractional deuteration of the wild type and a pentamutant (see below) PhTET2 protein (dPhTET2) was carried out in the ILL Deuteration Laboratory, Grenoble, France. Cells were grown at 30°C in minimal medium as described by Artero et al. (2005) using 85%(v/v) D2O and unlabelled glycerol as a carbon source. At an OD600 of about 10 for the wild type and of about 13 for the pentamutant construct, the high cell-density cultures were induced with 1 mM IPTG overnight. The final deuteration level was approximately 75% to yield a SANS contrast match point of 100% D2O. The resulting cell pellets were stored at −80°C until further use.
For purification, the pellets were thawed at room temperature and resuspended in 50 ml 50 mM Tris–HCl, 150 mM NaCl, 0.1% Triton X-100 pH 8.0. Next, 12.5 mg lysozyme (Euromedex), 2.5 mg DNase I grade II (Roche), 10 mg RNase (Roche), 50 mg Pefabloc SC (Roche) and 0.5 ml 2 M MgSO4 were added to the cell suspensions. Disruption of the cells was achieved by sonication in a Branson Sonifier 150 at 4°C. Five 30 s bursts at intensity 10 with intermediary pauses of 30 s were employed. The crude extract was then heated at 85°C for 15 min to eliminate most of the mesophilic proteins from the E. coli host and the lysates were clarified by centrifugation at 17 400g for 1 h. Supernatant concentrations were adjusted to 100 mM NaCl (dPhTET2) or 250 mM NaCl (hPhTET3), 20 mM Tris pH 7.5. The resulting extracts were loaded onto a 6 ml Resource Q column (GE Healthcare) previously equilibrated in 100 mM NaCl, 20 mM Tris–HCl pH 7.5. After washing with three column volumes (CV) of the equilibration buffer, bound proteins were eluted at 4 ml min−1 with a 20 CV linear salt gradient (0.1–0.35 M NaCl for dPhTET2 or 0.25–0.45 M NaCl for hPhTET3 in 20 mM Tris–HCl pH 7.5). For further purification, the proteins were loaded onto a Superose 6 size-exclusion column (GE Healthcare) equilibrated and run in 20 mM Tris–HCl, 150 mM NaCl pH 7.5.
2.2. De-oligomerization of native TET homododecamers and formation of heterododecamers
After the size-exclusion step of the purification, dodecameric dPhTET2 and hPhTET3 samples were dialyzed against a de-oligomerization buffer (50 mM CAPS, 20 mM NaCl, 20 mM EDTA pH 10). Aliquots of each sample were analyzed by size exclusion on a Superose 6 column (GE Healthcare) and on native polyacrylamide gels to control the oligomeric state of the samples after dialysis. According to the size-exclusion (Supplementary Fig. S11), both contained mostly TET dimers; the dPhTET2 sample contained less than 4% monomers and the hPhTET3 sample contained approximately 30% monomers. Therefore, less than 4% of the dimers present in solution can be dPhTET2–hPhTET3 heterodimers and such building blocks were therefore discarded in the modelling process, which was exclusively based on homodimeric building blocks (Figs. 1 and 2).
![[Figure 1]](wa5075fig1thm.gif) | Figure 1 Workflow of the SANS approach adopted to determine the quaternary arrangement of deuterated PhTET2 (black) and hydrogenated PhTET3 (grey) building blocks within the heterododecameric particles in solution. |
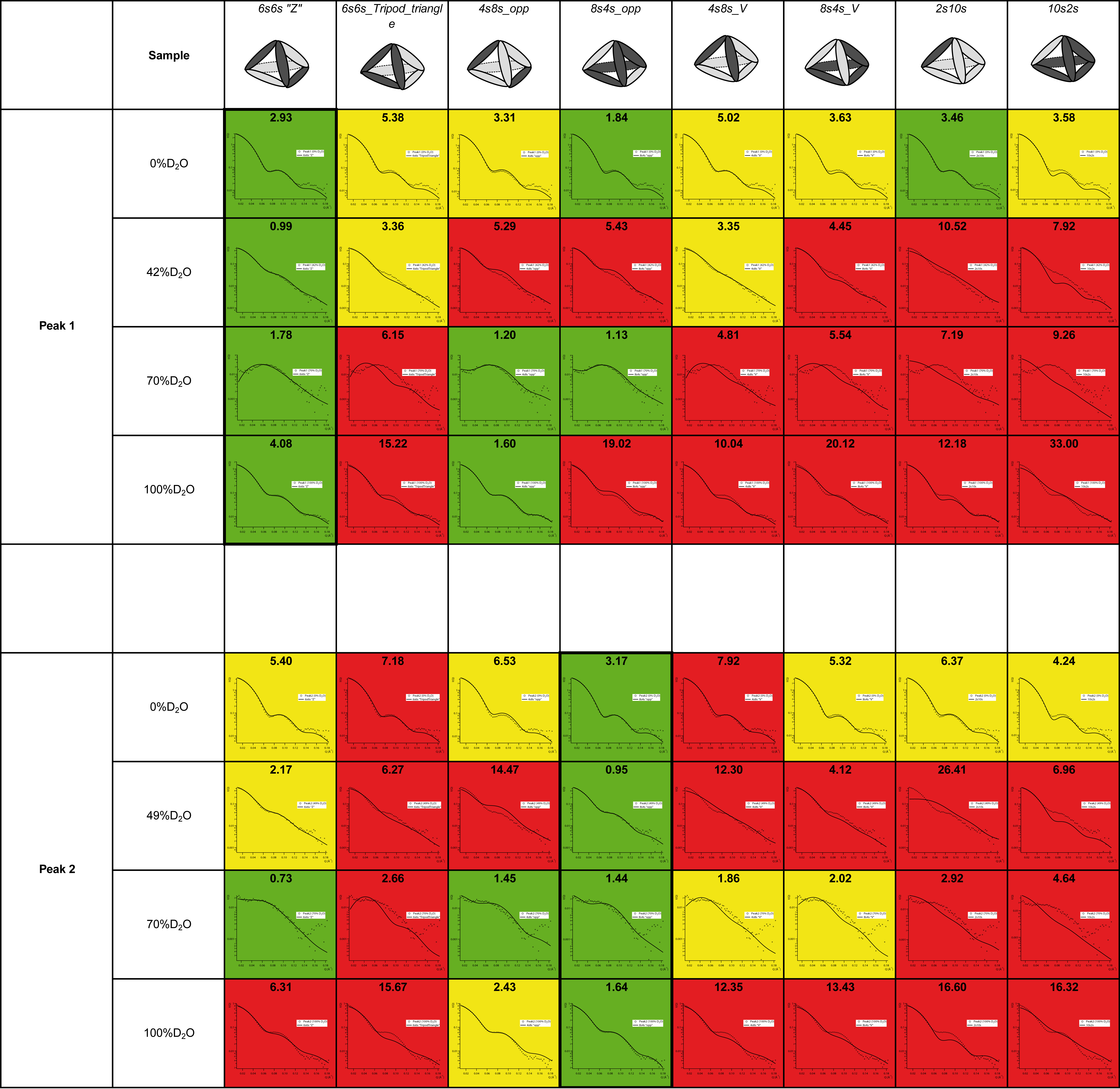
![[Figure 2]](wa5075fig2thm.gif) | Figure 2 Overview of the fits of several homodimeric models against the four SANS contrast scattering curves. Deuterated PhTET2 dimers are depicted by dark grey ellipsoids and hydrogenated PhTET3 dimers are depicted as light grey ellipsoids. Green indicates excellent fits against SANS data at the respective H2O/D2O contrast, yellow moderate agreement and red strong disagreement. The numbers indicate the χ2 values obtained with CRYSON. Peak 1 can only be fitted by one model in a satisfactory way (dPhTET2 6s:hPhTET3 6s = `Z'), while peak 2 can only be fitted by a single, distinct model (dPhTET2 8s:hPhTET3 4s = `opposite'). All other dodecameric models based on homodimeric building blocks can readily be ruled out. |
The de-oligomerized dPhTET2 and hPhTET3 samples were mixed together and dialyzed against re-oligomerization buffer (20 mM Tris–HCl, 150 mM NaCl, 2 mM CoCl2 pH 7.5). The re-oligomerized sample was analyzed on a Resource Q ion-exchange column (GE Healthcare) equilibrated in 150 mM NaCl, 20 mM Tris–HCl pH 7.5. After washing with 3 CV of the equilibration buffer, bound proteins were eluted at 4 ml min−1 with a 20 CV linear salt gradient (0.15–0.45 M NaCl in 20 mM Tris–HCl pH 7.5). Four peaks were obtained, two of which were predominant and contained a sufficient amount of protein for further characterization by SANS. According to their order of elution during the step, they were named peak 1 and peak 2. To increase the monodispersity of the samples, they were loaded onto a 1 ml Mono Q column (GE Healthcare) equilibrated in 150 mM NaCl, 20 mM Tris–HCl pH 7.5. After washing with 3 CV of the equilibration buffer, bound proteins were eluted at 1.5 ml min−1 with a 30 CV linear salt gradient (0.15–0.45 M NaCl in 20 mM Tris–HCl pH 7.5). They were then analyzed by (Superose 6) to control their final state and both were exclusively dodecameric (Supplementary Fig. S10).
2.3. SANS sample preparation, experimental details and data reduction
The samples corresponding to each peak from the final were split into two: one half was conserved in 100% H2O and the other half was equilibrated in 100% D2O. Buffer exchange (20 mM Tris–HCl, 150 mM NaCl pH 7.5 in 100% D2O or 100% H2O) was performed by dialysis overnight at 4°C. The D2O ratio of the different samples was then obtained by mixing the two solutions. 200 µl of protein solution was prepared at a concentration of 4.5 mg ml−1 at the following D2O ratios: 0, 42, 70 and 100% D2O. The D2O ratios were validated via their neutron transmission values. Apart from the 42% sample of peak 2, which was actually at 49% D2O, all samples were at the nominal ratio. Homododecameric hPhTET2 (42% D2O), dPhTET2 (42 and 100% D2O) and hPhTET3 (100% D2O) reference samples were prepared following the same protocol.
Protein concentrations were measured using the Bio-Rad protein-assay reagent (Bio-Rad) with bovine serum albumin as a standard. Correction factors were applied to pure PhTET2 and PhTET3 samples. These factors were calculated after determining the protein concentration of pure PhTET2 and PhTET3 samples by quantitative amino-acid analysis as described in Durá et al. (2009). For the concentration of pure hetero-oligomeric samples, a weighted correction factor was calculated taking into account the correction factors obtained for PhTET2 and PhTET3 and the number of monomers of these proteins in the heteromeric complexes. Therefore, all of the values reported in this paper refer to real protein concentrations. We estimate the accuracy of the concentration measurements to be about 30%.
SANS experiments were performed on the large dynamic range small-angle diffractometer D22 at the Institut Laue–Langevin (ILL), Grenoble, France. The incident wavelength was λ = 6 Å (Δλ/λ = 10%) at a single detector/collimator configuration (4 m/4 m) with a centred beam. All samples and buffers were prepared the night before the experiment as described above. The final sample volumes were adjusted to 160 µl and placed in Hellma 1 mm QS quartz cells. Boron, empty cell and a pure H2O sample were measured as references for the subsequent data treatment. In a first experiment, we verified the match points of dPhTET2 in 100% D2O and hPhTET2 in 42% D2O (Supplementary Fig. S9). The temperature of the remaining samples, including buffers and references, was increased in steps of 20°C from 20 to 80°C. Typical exposure times were 20 min for the reference samples, mixtures and buffers at each temperature step. In addition, the empty cell, H2O and boron were measured for 10 min at each temperature. As a result, all samples were kept at each temperature for 4 h. Transmissions were measured for 3 min at the lowest temperature (20°C). The two-dimensional data sets of all samples and buffers were reduced to one-dimensional curves using the standard ILL software (Gosh et al., 2006). Buffer intensities were subtracted from the respective sample intensities using PRIMUS from the ATSAS program suite (Konarev et al., 2003).
2.4. SANS data analysis and modelling
The model-free parameters I(0) (intensity scattered in the forward direction) and Rg (radius of gyration) were extracted from the SANS curves using the Guinier approximation (Guinier, 1939; Table 1). Pair-distance distribution functions p(r) were extracted using GNOM (Svergun, 1992) by imposing p(r = 0) = p(r = Dmax) = 0. Calibration of the molecular mass of the particles in solution was performed against water (Jacrot & Zaccai, 1981). The solvent-excluded volumes and scattering lengths of dPhTET2 and hPhTET3 monomers were calculated based on their amino-acid sequences (Jacrot, 1976), measured I(0), protein concentrations, transmissions and quartz-cell path lengths (Supplementary Table S1).
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Ab initio shapes of the PhTET2 and PhTET3 moieties within the heterododecamers (Supplementary Fig. S7) were calculated with MONSA (Svergun, 1999) using two phases (dPhTET2 and hPhTET3) according to the following parameters: monomer volumes VPhTET2 = VPhTET3 = 50 000 Å3 (6s:6s stoichiometry, where `s' designates a single subunit, i.e. a monomer), VPhTET2 = 66 700 Å3, VPhTET3 = 33 300 Å3 (8s:4s stoichiometry); the contrasts of dPhTET2 were 5.75, 3.34 (2.91) and −0.02 × 1010 cm−2 in 0, 42 (49) and 100% D2O and the contrasts of hPhTET3 were 2.30, −0.12 (−0.53) and −3.48 × 1010 cm−2 in 0, 42 (49) and 100% D2O. Connectivity of the two phases was not imposed.
For the pseudo-atomic models, PhTET2 and PhTET3 dodecameric models were generated from the crystal structures (PDB entries 1y0r and 2wzn, respectively; Borissenko & Groll, 2005; Durá et al., 2009). Indeed, both crystal structures present a monomer within the asymmetric unit, so generating the dodecameric models relied on only the same symmetry operators. Both dodecameric models were adjusted to yield best fits (Fig. 1, top; Supplementary Fig. S12) against the respective SANS curves of the homododecameric reference samples dPhTET2 12s (42% D2O) and hPhTET3 12s (100% D2O) using CRYSON (Svergun et al., 1995, 1998). In particular, the missing parts of the respective crystal structures were added manually with Coot (Emsley & Cowtan, 2004). In the case of the PhTET2 model, addition of the missing part of the N-terminus (residues 1–5) as well as the internal loop (residues 120–132) led to a good fit of the dPhTET2 12 s SANS reference curve in 42% D2O (Fig. 1, top; Supplementary Fig. S12). As a basic unit, dimeric building blocks were chosen (in agreement with previous SAXS data; Appolaire et al., 2013) and generated with the symmetry operator prior to generating the whole dodecameric model. In the case of PhTET3, addition of the N-terminal part (residues 1–7) as well as the internal loop (residues 128–136) led to a nonsatisfactory fit of the hPhTET3 12s SANS reference curve in 100% D2O. Consequently, the PhTET3 dimeric building bock was generated using a manual rigid body to slightly change the relative position of one monomer with respect to the second monomer within the dimeric building block. In this way and by adding the N-terminal part, a dodecameric model was generated with a reasonably good fit to the SANS reference curve (Fig. 1, top; Supplementary Fig. S12). An overview comparing both of the dodecameric TET particles before and after the modifications as well as with each other is provided in Supplementary Fig. S11.
In order to facilitate the generation of heterododecameric hybrids, prior to dodecamer generation PhTET2 and PhTET3 dimeric models were superimposed in Coot. A library of heterododecameric hybrids was then created by editing the PDB files. The library exhaustively covered all dodecameric models based on homodimeric PhTET2 and PhTET3 building blocks (Fig. 2). All structures from the library were scored in a least χ2 fit against the SANS data in 0, 42 (49), 70 and 100% D2O for peak 1 and peak 2 using CRYSON. Fits were classified as excellent, moderate and bad for each model and contrast (Fig. 2) and were colour-coded green, yellow and red, respectively. A complete version of Fig. 2, including the individual fit curves, is provided as Supplementary Fig. S13. It illustrates that both χ2 values and visual criteria (good overall superposition of back-calculated and experimental curves) should be considered when discriminating between different models.
3. Results
3.1. Purification of PhTET2–PhTET3 hetero-oligomers
In the present study, we induced de-oligomerization of dodecameric deuterium-labelled PhTET2 (`dPhTET2') and dodecameric hydrogenated PhTET3 (`hPhTET3') into dimers. Subsequently, hetero-oligomers were generated by mixing the two dimer populations and inducing their re-oligomerization. Since PhTET2 and PhTET3 do not have the same external surface charges, their elution volumes are very different when analyzed appropriately by ion-exchange chromatography. Consequently, the different hetero-oligomeric assemblies can be separated by using an adequate ion-exchange chromatographic column run with a suitable salt gradient. After a first chromatographic step on a Resource Q column, two major (and several minor) peaks were observed (Fig. 3a). In order to better separate the different hetero-oligomers, a second strong ion-exchange chromatography was performed on a Mono Q column (Fig. 3b). (The fractions recovered from the Resource Q for the Mono Q are shaded in grey in Fig. 3a.) The protein concentrations and monodispersity of the two major peaks (`peak 1' and `peak 2') were high enough for subsequent SANS experiments.
![[Figure 3]](wa5075fig3thm.gif) | Figure 3 Purification of heterododecamers. The peaks are represented using the absorbance (A280 nm) as a function of the conductivity (mS cm−1). (a) Ion-exchange chromatography (Resource Q) of the total sample after the re-oligomerization process. The shaded areas correspond to the fractions grouped together for successive Mono Q purification. (b) Ion-exchange chromatography (Mono Q) of the two major peaks after the first Resource Q step. The shaded areas correspond to the fractions grouped together for size-exclusion chromatography on a Superose 6 column (Supplementary Fig. S10). |
3.2. SANS data reveal two extremely stable, heterododecameric dPhTET2–hPhTET3 architectures of different stoichiometry and quaternary structure
The samples from both peaks yielded very specific and distinct SANS curves (Fig. 4a) and model-free parameters I(0) and Rg (Table 1) at the four contrast conditions. At 0% D2O both resemble the homododecameric reference curves (Supplementary Fig. S6) most since both dPhTET2 and hPhTET3 moieties have positive contrast with respect to the solvent (Supplementary Fig. S4). The 70% D2O data have the lowest relative intensities since the hydrogenated and deuterated building blocks are of opposite contrast and result in a decrease in the overall scattered intensities. At 42 (49) and 100% D2O the scattered signals are almost exclusively owing to the deuterated and hydrogenated building blocks of the particles, respectively. Since small but significant variations of the curves at 0, 42 (49), 70 and 100% D2O are observed for both peaks, it can be concluded that the geometric arrangement of dPhTET2 and hPhTET3 building blocks are different within the particles constituting peak 1 and peak 2.
![[Figure 4]](wa5075fig4thm.gif) | Figure 4 SANS curves and p(r) functions of peak 1 and peak 2. (a) SANS curves at four different contrast conditions at 20°C. All data sets are drawn without applying scaling. The total protein concentrations of all samples were identical (4.5 mg ml−1). Guinier fits to the 0, 42 (49) and 100% D2O data are shown as an inset (the 70% D2O data were not fitted using the Guinier approach owing to their elevated noise level). (b) Pair-distance distribution functions p(r) of the SANS data in arbitrary units generated with GNOM (Svergun, 1992 ). The two 0% D2O data sets are very similar (cf. Supplementary Fig. S6). The 0, 42, 49 and 100% D2O data are normalized to the second peak and the 70% D2O data to the first peak (using a different scaling factor for clarity). |
An intriguing feature is the evolution of the I(0) intensities as a function of contrast: while particle 2 (peak 2) scatters more strongly in the forward direction at 0, 42 and 70% D2O than particle 1 (peak 1), the situation is the inverse at 100% D2O. Since I(0), at a given total protein concentration, is proportional to the contrast integrated over the whole particle volume (Svergun & Koch, 2002), these findings indicate that particle 2 contains more deuterated building blocks than particle 1. Interestingly, these findings imply that at least two stoichiometrically different architectures are possible for heterododecameric TET particles in vitro. A calibration of the molecular masses against water (Jacrot & Zaccai, 1981) of the 0% D2O data yielded a dPhTET2:hPhTET3 stoichiometry of 6:6 for particle 1 and of 8:4 for particle 2 (Supplementary Table S1). Importantly, both hetero-oligomeric TET particles display extreme structural stability at high temperatures, with the SANS curves being virtually identical at 20 and 80°C (Supplementary Fig. S2). The same stability was found for mixtures of preformed homododecameric dPhTET2 and hPhTET2 particles for both the wild type and a pentamutant variant designed to weaken the oligomerization interface (Supplementary Fig. S3). Importantly, these data rule out a dynamic equilibrium between dodecameric assemblies and putative smaller oligomers in solution under our experimental conditions. The quality of the matching of hydrogenated and deuterated PhTET2 was checked by measuring dodecameric hPhTET2 and dPhTET2 particles in 42 and 100% D2O, respectively. The results (Supplementary Fig. S9) indicated that the matching conditions were excellent, i.e. contributions from hydrogenated moieties can be neglected at 42% D2O and those of deuterated moieties at 100% D2O.
The pair-distance distribution functions p(r) extracted with GNOM (Svergun, 1992) from the SANS curves at the four contrast conditions revealed a wealth of real-space information on the relative positions of deuterated and hydrogenated building blocks within the two particles (Fig. 4b). The back-fitted (regularized) scattering curves are shown in Supplementary Fig. S5 and show excellent agreement with the experimental data. An overview of the numerical values of Rg and Dmax is provided in Table 1.
At 0% D2O both dPhTET2 and hPhTET3 building blocks have positive contrast (of different amplitude) with respect to the solvent (Supplementary Fig. S4). The p(r) functions from peak 1 and peak 2 are therefore almost identical, indicating that both particles possess a very similar overall shape (envelope) comparable to the dPhTET2 and hPhTET3 homododecameric references (Supplementary Fig. S6). The maxima of the leaning bell curves are shifted from the centre to larger distances and then drop rapidly to zero (a hallmark of hollow, globular particles; Koch et al., 2003) and are in good agreement with the overall topology of dodecameric TET particles. The Dmax of 130 Å is also in excellent agreement with the expected PhTET dimensions from electron microscopy (Schoehn et al., 2006).
At 42 (49)% D2O the p(r) functions are almost exclusively owing to deuterated moieties and therefore represent the internal arrangement of dPhTET2 building blocks within the dodecameric edifices. In both cases a broadened central peak is observed. A tendency to split up into a bimodal pattern is observed for both particles: symmetric for particle 1 and asymmetric for particle 2. The observed bimodal p(r) patterns can be interpreted in terms of several compact, deuterated moieties positioned at a finite distance from each other, being either loosely connected or separated by small gaps. The 100% D2O data represent the complementary contrast condition, now with the hydrogenated building blocks (hPhTET3) almost exclusively accounting for the p(r) patterns. Again, bimodal patterns are observed. Interestingly, the separation of both humps is more pronounced for particle 2, indicating that its respective hPhTET3 dimers are, on average, more widely separated than those in particle 1.
At 70% D2O hPhTET3 and dPhTET2 building blocks have negative and positive contrast, respectively (Supplementary Fig. S4). Since p(r) functions contain products of pairs of volume elements weighted by their respective contrast, positive and negative regions can be observed in such cases (Glatter & Kratky, 1982): positive p(r) values occur if volume elements separated by the distance r are predominantly of identical contrast (positive–positive or negative–negative) and negative values are observed if volume elements of opposite contrast (positive–negative or negative–positive) dominate at this distance. The positive peak at short distances (r ≃ 20 Å) therefore belongs to volume elements within dPhTET2 (or hPhTET3) dimeric building blocks. At intermediate distances (r ≃ 60 Å) volume elements of opposite contrast (dPhTET2–hPhTET3 pairs) prevail, yielding a minimum, while at large distances (r ≃ 100–110 Å) pairs of volume elements with the same sign again dominate. Intriguingly, at 70% D2O the minimum at 60 Å is more pronounced for particle 1 than particle 2, indicating that in the former the dPhTET2 and hPhTET3 dimers are distributed more symmetrically (in number and geometry).
3.3. Pseudo-atomic models of PhTET2–PhTET3 complexes using SANS contrast-variation data
Fig. 1 illustrates the workflow of our approach to determine pseudo-atomic, quaternary structures of the heterododecameric PhTET2–PhTET3 particles. As a first step, homododecameric (`12s') reference structures of dPhTET2 and hPhTET3 were measured by SANS at 42 and 100% D2O, respectively. PhTET2 and PhTET3 dodecameric crystal structures (PDB entries 1y0r and 2wzn, respectively) were slightly modified to match these SANS reference data at `low resolution' (Fig. 1, top) and were subsequently used to construct a library of heterododecameric models of variable stoichiometry and geometry (Figs. 1 and 2, Supplementary Fig. S13). All structures from the library were scored against the SANS data from peak 1 and peak 2 at 0, 42 (49), 70 and 100% D2O. A colour code (green, yellow and red) was used to design excellent, moderate and unacceptable fits. Only one architecture was able to satisfy all contrast conditions simultaneously for each peak and all others could readily be rejected (Fig. 2, Supplementary Fig. S13). Peak 1 was only fitted by a dPhTET2 6s/hPhTET3 6s architecture with the two hexamer (`6s') moieties arranged in an intertwined, double Z-shaped, clamp-like form (Fig. 5a). Peak 2 was only fitted by a dPhTET2 8s/hPhTET3 4s model with the PhTET3 moiety composed of two homodimers (`4s'; four monomers) situated at opposite ridges of the dodecameric particle (Fig. 5b).
![[Figure 5]](wa5075fig5thm.gif) | Figure 5 SANS data and best models for peak 1 and peak 2: the two architectures (`Z' and `opposite') that are in best agreement with the SANS data of peak 1 (a) and peak 2 (b), along with their respective fits at four different contrasts. Elongated ellipsoids represent dPhTET2 (dark grey) and hPhTET3 (light grey) homodimers. Intriguingly, each tetrahedral apex brings together catalytic sites of different substrate specificity. |
In addition, and in complement, to the pseudo-atomic models, the multiphase ab initio program MONSA (Svergun, 1999) was used to calculate low-resolution envelopes of the PhTET2 and PhTET3 moieties within the heterododecameric particles of peak 1 and peak 2. A comparison of the low-resolution models with the pseudo-atomic models is shown in Supplementary Fig. S7. The ab initio shapes confirm, independently of the pseudo-atomic models, that PhTET2 and PhTET3 are organized as a symmetric architecture of two intertwined Z-shapes in particle 1 and that PhTET2 is organized as two homodimers positioned on opposite ridges in particle 2.
3.4. The homododecameric PhTET2 complex is extremely stable in solution and is not in dynamic equilibrium with free oligomeric forms of lower molecular mass
As an additional stability control of the PhTET quaternary structure and to test the presence of putative equilibria between preformed dodecameric particles with smaller oligomeric particles in solution, we measured mixtures of pre-formed homododecameric deuterated and hydrogenated PhTET2 proteins (dPhTET2 `12s' and hPhTET2 `12s') by monitoring their SANS curves as a function of temperature and exposure time. The experiments were performed on both the wild-type protein and a pentamutant (R217S, R220S, F224S, H248S and I292A) with the mutations situated at the interface between the dimers and designed to slow the oligomerization process (Appolaire et al., 2013). For the wild-type and the mutated PhTET2, the following samples were prepared: hPhTET2 (42% D2O), dPhTET2 (42% D2O), hPhTET2 (100% D2O) and dPhTET2 (100% D2O). Different mixtures of the above samples were prepared by mixing 1:1 volume fractions of these solutions.
The SANS curves of isolated hPhTET2 (100% D2O) and dPhTET2 (42% D2O) reference particles (Supplementary Fig. S3) revealed the typical pattern of the dodecameric edifice as observed in previous SANS experiments (Durá et al., 2009). Supplementary Fig. S3(a) shows a superposition of the scattering curves of wild-type hPhTET2 in 100% D2O at t = 0 (20°C) and of a 1:1 mixture of dPhTET2 and hPhTET2 in 100% D2O after 4 h at 80°C, both normalized to hPhTET2 concentration. Supplementary Fig. S3(b) shows the respective data sets of a 1:1 dPhTET2/hPhTET2 mixture at 42% D2O. Interestingly, the scattering patterns of both mixtures after 4 h at 80°C are identical to those of their respective dodecameric PhTET2 references at 20°C regarding radii of gyration, position of the side maxima and minima and I(0) intensities. These very clear results indicate that no exchange of lower oligomeric building blocks between the hydrogenated and deuterated dodecamers had occurred in solution on the temperature and time scales of our SANS experiments. These experiments confirm the great stability of both the wild-type and pentamutant dodecameric particles once they are formed in solution.
4. Discussion
Large and symmetric complexes of multiple copies of a single or a few similar building blocks play essential roles in the life cycle of biological cells (Matthews & Sunde, 2012; Griffin & Gerrard, 2012; Sauer & Baker, 2011; Snider & Houry, 2008; Marianayagam et al., 2004). If crystal structures are not available, they represent a formidable challenge for many structural biology techniques: for NMR owing to their size and for SAXS or EM because it is very difficult to position individual subunits within the assembled complexes. The heterododecameric PhTET2–PhTET3 complexes studied here are highly symmetric and we applied a powerful combination of SANS, deuterium labelling and contrast variation to obtain unique insights into the oligomeric organization of their quaternary architectures. In contrast to EM or SAXS, the neutron scattering lengths for hydrogen and deuterium differ significantly and are of opposite sign (Jacrot, 1976). SANS can therefore focus specifically on D- or H-labelled partners within a reconstituted complex. Using this approach, we were able to eliminate a multitude of potential stoichiometries and geometric arrangements very efficiently and to clearly identify a single, specific architecture of the PhTET2 and PhTET3 moieties within the final heterododecameric complexes from peak 1 and peak 2 (Fig. 2, Supplementary Fig. S13).
Previously, a combination of site-directed mutagenesis, SAXS, AUC and EM allowed us to propose an assembly mechanism in which three dimers associate into a hexameric precursor prior to the formation of TET dodecamers (Appolaire et al., 2013). This model accounts well for the very limited number of hetero-oligomeric TET complexes that we have identified in the present work by ion-exchange chromatography after metal-induced re-oligomerization of a mixture of PhTET2 and PhTET3 dimers (Fig. 3). This strategy was possible because the PhTET2 and PhTET3 partners behave differently on strong ion-exchange chromatography, allowing the separation of different hetero-oligomeric complexes as a function of their PhTET2:PhTET3 ratio. The SANS study described here provides direct insight into this assembly model. Indeed, the topology of the PhTET2 and PhTET3 moieties within the two main hetero-oligomeric species observed are in excellent agreement with an assembly process that involves a hexameric intermediate (Fig. 6, Supplementary Fig. S8), as suggested by a previous study combining SAXS, EM and AUC (Appolaire et al., 2013).
![[Figure 6]](wa5075fig6thm.gif) | Figure 6 Assembly pathway for the `Z' dPhTET2 6s:hPhTET3 6s heterododecamer (peak 1): the observation of a specific internal hexamer–hexamer topology by SANS, together with the exclusion of alternative topologies, points to a specific assembly pathway involving intertwined heterohexamers. Thick continuous and thin broken arrows represent strong and weak pathways for the dynamic equilibrium between respective states. An alternative assembly mode is presented in Supplementary Fig. S8. |
Each of the TET apices defines a catalytic subcompartment in which three active sites are located on the same plane. Moreover, a nonprocessive mode of action has been reported for the three TET proteins from P. horikoshii (Durá et al., 2005, 2009; Schoehn et al., 2006). Therefore, after the cleavage of the N-terminal amino acid the polypeptide substrate is released from the active site and is free to interact with one of the two other catalytic pockets. In the present study we showed that, unexpectedly, TET dodecameric quaternary structures follow a highly organized pathway when self-assembling in vitro. This results in a limited number of architectures with a single type of apex composition, systematically combining PhTET2 and PhTET3 active sites with different and complementary substrate specificities (Fig. 6), suggesting that the highly controlled oligomerization process of the PhTET2–PhTET3 complex is to optimize its peptide-degradation efficiency.
Finally, our work shows that SANS experiments allow the respective positions of the different subunits to be very efficiently specified in the case of a large symmetrical pseudo-homomeric complex, information that is very difficult, if not impossible, to obtain using most biophysical techniques. In conclusion, such a SANS approach could be very efficient to better understand the assembly pathway of other large, symmetric, pseudo-homomeric complexes such as the proteasome (Sahara et al., 2014), chaperonins (Kim et al., 2013) or AAA-ATPases (Bar-Nun & Glickman, 2012). Our structural insights raise intriguing questions on the underlying regulatory mechanisms controlling the assembly process of such biomacromolecular complexes, a field of research that has remained poorly explored to date but might offer important perspectives for biomedical research and drug development in the future.
Supporting information
Supplementary Figs. S1-S12 and Supplementary Table S1. DOI: https://doi.org/10.1107/S1399004714018446/wa5075sup1.pdf
High resolution Supplementary Fig. S13. DOI: https://doi.org/10.1107/S1399004714018446/wa5075sup2.png
Acknowledgements
We acknowledge financial support from the Agence Nationale de la Recherche (grants `MacroTET' BLAN-07-3 204002, `Archelyse' ANR-12-BSV8-0019-01 and `HYDROSAS' ANR-11-JSV5-003-01). AA was supported by a PhD scholarship from the French Ministry for Research and Technology. MAD and MC were supported by a French National Research Agency postdoctoral fellowship. We would like to thank the ILL for SANS BAG beamtime on D22 and P. Callow and A. Martel for help with the set-up of the SANS instrument. MM and MH acknowledge EPSRC support (EP/C015452/1) to V. T. Forsyth (Keele University, England) for the creation of the Deuteration Laboratory within ILL's Life Sciences Group and from the EU under contract RII3-CT-2003-505925.
References
 Appolaire, A., Rosenbaum, E., Durá, M. A., Colombo, M., Marty, V., Savoye, M. N., Godfroy, A., Schoehn, G., Girard, E., Gabel, F. & Franzetti, B. (2013). J. Biol. Chem. 288, 22542–22554. Web of Science CrossRef CAS PubMed Google Scholar
Appolaire, A., Rosenbaum, E., Durá, M. A., Colombo, M., Marty, V., Savoye, M. N., Godfroy, A., Schoehn, G., Girard, E., Gabel, F. & Franzetti, B. (2013). J. Biol. Chem. 288, 22542–22554. Web of Science CrossRef CAS PubMed Google Scholar
Artero, J.-B., Härtlein, M., McSweeney, S. & Timmins, P. (2005). Acta Cryst. D61, 1541–1549. Web of Science CrossRef CAS IUCr Journals Google Scholar
Bar-Nun, S. & Glickman, M. H. (2012). Biochim. Biophys. Acta, 1823, 67–82. CAS PubMed Google Scholar
Borissenko, L. & Groll, M. (2005). J. Mol. Biol. 346, 1207–1219. Web of Science CrossRef PubMed CAS Google Scholar
Chaikuad, A., Pilka, E. S., De Riso, A., von Delft, F., Kavanagh, K. L., Vénien-Bryan, C., Oppermann, U. & Yue, W. W. (2012). BMC Struct. Biol. 12, 14. Google Scholar
Chen, Y., Farquhar, E. R., Chance, M. R., Palczewski, K. & Kiser, P. D. (2012). J. Biol. Chem. 287, 13356–13370. Web of Science CrossRef CAS PubMed Google Scholar
Durá, M. A., Receveur-Brechot, V., Andrieu, J.-P., Ebel, C., Schoehn, G., Roussel, A. & Franzetti, B. (2005). Biochemistry, 44, 3477–3486. PubMed Google Scholar
Durá, M. A., Rosenbaum, E., Larabi, A., Gabel, F., Vellieux, F. M. & Franzetti, B. (2009). Mol. Microbiol. 72, 26–40. PubMed Google Scholar
Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. Web of Science CrossRef CAS IUCr Journals Google Scholar
Franzetti, B., Schoehn, G., Hernandez, J. F., Jaquinod, M., Ruigrok, R. W. & Zaccai, G. (2002). EMBO J. 21, 2132–2138. Web of Science CrossRef PubMed CAS Google Scholar
Glatter, O. & Kratky, O. (1982). Small Angle X-ray Scattering. New York: Academic Press. Google Scholar
Gosh, R. E., Egelhaaf, S. U. & Rennie, A. R. (2006). A Computing Guide for Small-angle Scattering Experiments. Grenoble: Institut Laue–Langevin. Google Scholar
Griffin, M. & Gerrard, J. (2012). Protein Dimerisation and Oligomerisation in Biology, edited by J. Matthews, pp. 74–90. New York: Springer. Google Scholar
Guinier, A. (1939). Ann. Phys. (Paris), 12, 161–236. CAS Google Scholar
Jacrot, B. (1976). Rep. Prog. Phys. 39, 911–953. CrossRef CAS Web of Science Google Scholar
Jacrot, B. & Zaccai, G. (1981). Biopolymers, 20, 2413–2426. CrossRef CAS Web of Science Google Scholar
Kim, D., San, B. H., Moh, S. H., Park, H., Kim, D. Y., Lee, S. & Kim, K. K. (2010). Biochem. Biophys. Res. Commun. 391, 431–436. Web of Science CrossRef PubMed CAS Google Scholar
Kim, Y. E., Hipp, M. S., Bracher, A., Hayer-Hartl, M. & Hartl, F. U. (2013). Annu. Rev. Biochem. 82, 323–355. Web of Science CrossRef CAS PubMed Google Scholar
Koch, M. H. J., Vachette, P. & Svergun, D. I. (2003). Q. Rev. Biophys. 36, 147–227. Web of Science CrossRef PubMed CAS Google Scholar
Konarev, P. V., Volkov, V. V., Sokolova, A. V., Koch, M. H. J. & Svergun, D. I. (2003). J. Appl. Cryst. 36, 1277–1282. Web of Science CrossRef CAS IUCr Journals Google Scholar
Marianayagam, N. J., Sunde, M. & Matthews, J. M. (2004). Trends Biochem. Sci. 29, 618–625. Web of Science CrossRef PubMed CAS Google Scholar
Matthews, J. & Sunde, M. (2012). Protein Dimerisation and Oligomerisation in Biology, edited by J. Matthews, pp. 1–18. New York: Springer. Google Scholar
Rawlings, N. D., Waller, M., Barrett, A. J. & Bateman, A. (2014). Nucleic Acids Res. 42, D503–D509. Web of Science CrossRef CAS PubMed Google Scholar
Rosenbaum, E., Ferruit, M., Durá, M. A. & Franzetti, B. (2011). Biochim. Biophys. Acta, 1814, 1289–1294. Web of Science CrossRef CAS PubMed Google Scholar
Rosenbaum, E., Gabel, F., Durá, M. A., Finet, S., Cléry-Barraud, C., Masson, P. & Franzetti, B. (2012). Arch. Biochem. Biophys. 517, 104–110. Web of Science CrossRef CAS PubMed Google Scholar
Russo, S. & Baumann, U. (2004). J. Biol. Chem. 279, 51275–51281. Web of Science CrossRef PubMed CAS Google Scholar
Sahara, K., Kogleck, L., Yashiroda, H. & Murata, S. (2014). Adv. Biol. Regul. 54, 51–58. CrossRef CAS PubMed Google Scholar
Sauer, R. T. & Baker, T. A. (2011). Annu. Rev. Biochem. 80, 587–612. Web of Science CrossRef CAS PubMed Google Scholar
Schoehn, G., Vellieux, F. M. D., Durá, M. A., Receveur-Bréchot, V., Fabry, C. M. S., Ruigrok, R. W. H., Ebel, C., Roussel, A. & Franzetti, B. (2006). J. Biol. Chem. 281, 36327–36337. Web of Science CrossRef PubMed CAS Google Scholar
Snider, J. & Houry, W. A. (2008). Biochem. Soc. Trans. 36, 72–77. Web of Science CrossRef PubMed CAS Google Scholar
Svergun, D. I. (1992). J. Appl. Cryst. 25, 495–503. CrossRef Web of Science IUCr Journals Google Scholar
Svergun, D. I. (1999). Biophys. J. 76, 2879–2886. Web of Science CrossRef PubMed CAS Google Scholar
Svergun, D., Barberato, C. & Koch, M. H. J. (1995). J. Appl. Cryst. 28, 768–773. CrossRef CAS Web of Science IUCr Journals Google Scholar
Svergun, D. I. & Koch, M. H. J. (2002). Rep. Prog. Phys. 66, 1735–1782. Web of Science CrossRef Google Scholar
Svergun, D. I., Richard, S., Koch, M. H. J., Sayers, Z., Kuprin, S. & Zaccai, G. (1998). Proc. Natl Acad. Sci. USA, 95, 2267–2272. Web of Science CrossRef CAS PubMed Google Scholar
This is an open-access article distributed under the terms of the Creative Commons Attribution (CC-BY) Licence, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are cited.
| STRUCTURAL BIOLOGY |
access



{kind=link}