

 journal menu
journal menu





issue contents
May 2015 issue

Cover illustration: The solvent component of macromolecular crystals (Weichenberger et al., p. 1023). Given favourable kinetics, macromolecules can self-assemble from a metastable, supersaturated solution into crystals, a periodic network of macromolecules connected by weak but specific intermolecular interactions. On average, the mother liquor or solvent and its constituents occupy about 50% of a macromolecular crystal as in this example of a simple P4 crystal structure (PDB entry 2on8).
feature articles





Free 

On average, the mother liquor or solvent and its constituents occupy about 50% of a macromolecular crystal. Ordered as well as disordered solvent components need to be accurately accounted for in modelling and refinement, often with considerable complexity.
research papers

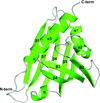
The crystal structure of ginseng major latex-like protein 151, its recognition of lysophosphatidic acid (LPA) and the activation of G protein-coupled LPA receptors by its complex with LPA are reported.

A method is presented to quantitatively assess the inherent ambiguity of three-dimensional shape reconstruction from a given small-angle scattering pattern.
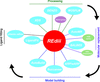
The REdiii software pipeline allows fully automated crystal structure solution by embedding existing tools into a self-managing workflow.
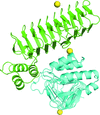
Ligand-free and UDP-N-acetylglucosamine-bound crystal structures of B. fragilis UDP-N-acetylglucosamine acyltransferase (BfLpxA) are reported.
ASSP is a freely available algorithm for secondary-structure assignment in protein structures. The algorithm is an extension of HELANAL-Plus and can be accessed at http://nucleix.mbu.iisc.ernet.in/assp/index.html.
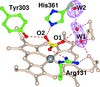
X-ray-induced substrate (nitrite and sulfite) transformations are studied for cytochrome c nitrite reductase from T. nitratireducens.
PDB references: TvNiR, free form, low-dose data set, 4q0t; medium-dose data set, 4q17; high-dose data set, 4q1o; nitrite complex, low-dose data set, 4l38; first medium-dose data set, 4l3x; second medium-dose data set, 4l3z; high-dose data set (NO complex), 4l3y; sulfite complex, low-dose data set, 4q4u; medium-dose data set, 4q5c; high-dose data set, 4q5b
De novo structure determination by sulfur SAD (S-SAD) was achieved despite a low anomalous signal, radiation damage and low redundancy. Based on this case and statistics from S-SAD structures in the PDB threshold values for successful S-SAD structure determination are discussed.
PDB reference: PilBac1, 4us7

MERS-CoV 3CLpro has been crystallized and X-ray crystal structures have been solved in three different crystal forms representing the free enzyme structure and the enzyme–product complex.
The use of the Thermofluor assay for membrane proteins was explored by testing five different detergents with common fluorescent probes. The dye 1-anilinonaphthalene-8-sulfonic acid was identified as a valuable tool when working with detergents and the membrane protein NhaA.

The fishelectins (FELs) are a protein family first identified in carp eggs and now recognized to be present in fish and amphibians. Here, the X-ray structure of carp FEL is reported both in its apo form and bound to the α- and β-anomers of N-acetylglucosamine. This is the first experimentally determined structure of a member of this protein family.

The unique structure of thermostable meso-diaminopimelate dehydrogenase from U. thermosphaericus was determined and compared with those of corresponding enzymes from other sources.
Open access
access
A method to automatically identify possible elemental ions in X-ray crystal structures has been extended to use support vector machine (SVM) classifiers trained on selected structures in the PDB, with significantly improved sensitivity over manually encoded heuristics.
Open access
access
The purified putative betaine aldehyde dehydrogenase SACOL2628 from the early methicillin-resistant S. aureus COL has betaine aldehyde dehydrogenase activity and is structurally similar to aldehyde dehydrogenases.
PDB references: betaine aldehyde dehydrogenase, 4mpb; complex with NAD+, 4mpy; complex with NAD+ with BME-free Cys289, 4nea; with BME-free Cys289, 4nu9; with BME-modified Cys289 and PEG molecule in active site, 4qto; G234S mutant, in complex with NAD+ with BME-free Cys289, 4qn2; G234S mutant, with BME-modified Cys289, 4q92; G234S mutant, with BME-free sulfinic acid form of Cys289, 4qje

The 2.55 Å resolution crystal structure of S100B in complex with a 15-amino-acid peptide derived from the receptor for advanced glycation end products is reported.
PDB reference: Ca2+-S100B, complex with human RAGE-derived peptide, 4xyn
Open access
access
A raster scanning serial protein crystallography approach is presented, that consumes as low ∼200–700 nl of sedimented crystals. New serial data pre-analysis software, NanoPeakCell, is introduced.
Open access
access
A helix swap involving the fifth helix between two adjacently bound Tah1 molecules restores the normal binding environment of the conserved MEEVD peptide of Hsp90. Dimerization also explains how other monomeric TPR-domain proteins are excluded from forming inappropriate mixed co-chaperone complexes with Hsp90 and Tah1.
PDB reference: Tah1, complex with Hsp90 peptide, 4cgq
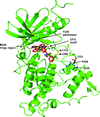
The crystal structure of DYRK1A in complex with PKC412 and data for the binding of PKC412 to DYRK1A/1B are reported. This structure confirms the unique ability of DYRK kinases to form a disulfide bridge between the catalytic loop and the activation segment.
PDB reference: DYRK1A in complex with PKC412, 4nct
The structure of S. aureus homoserine dehydrogenase at different pH conditions provides a basis for understanding the role of a lysine residue (Lys105) in the hydride-transfer step of the reaction mechanism.







