

 journal menu
journal menu





issue contents
December 2015 issue

Cover illustration: Human cytochrome P450 embedded in a phospholipid nanodisc (Skar-Gislinge et al., p. 2412).
research papers







A study using the multimeric enzyme tryptophanase as a model and crystal structure analysis suggests a structural view of protein–protein interaction and of the mechanism of dissociation at the molecular level.
PDB reference: tryptophanase, 5d8g
The structure and the assembly process of the photosynthetic GAPDH–CP12–PRK complex and its single components, i.e. PRK and the GADPH–CP12 complex, from A. thaliana have been investigated by small-angle X-ray scattering.
Open  access
access
access
The crystal structure of a P. aeruginosa DsbA1 variant is more suitable for fragment-based lead discovery efforts to identify inhibitors of this antimicrobial drug target. In the reported structures the active site of the protein can simultaneously bind multiple ligands introduced in the crystallization solution or via soaking.
12 crystallographic structures of T. thermophilus HB27 multicopper oxidase in holo, apo and Hg-bound forms and with different absorbed X-ray doses are reported, revealing the first structural evidence for the proton-relay mechanism in the X-ray-induced reduction of O2 to 2H2O at the trinuclear copper cluster of the enzyme. Different O2-reduction states and a total depletion of T2Cu at radiation doses higher than 0.2 MGy were also observed.
A combined ab initio and rigid-body approach has been developed for small-angle scattering analysis. This provides a previously inaccessible insight into the low-resolution structure of the human cytochrome P450 CYP3A4 when embedded in nanodiscs mimicking a native membrane.
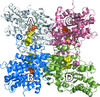
S-Adenosyl-L-homocysteine hydrolase from a nitrogen-fixing bacterium was crystallized as a biologically relevant homotetramer composed of subunits in different conformational states. While three of them are in the Ado product-bound closed conformation, the fourth is empty and open.

The crystal structure of the GH42 β-galactosidase Gan42B from G. stearothermophilus has been determined in its wild-type form (2.45 Å resolution) and its nucleophile catalytic mutant (E323A; 2.50 Å resolution). These structures show that the Gan42B protein is a homotrimer in which the three active sites are formed at the interfaces between each pair of monomers that build it, all facing a cone-shaped internal cavity.
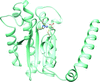
The kinetic characterization and the crystal structure of the recombinant type II β-CA from V. cholerae determined to a resolution of 1.9 Å with a closed active-site configuration are reported.
PDB reference: β-carbonic anhydrase, 5cxk
Open access
access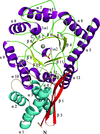
Non-ligand-bound and PEP-bound structures of S. aureus enolase (Sa_enolase) were solved, and catalytic loop 1 in the PEP-bound structure was found to show both `open' and `closed' conformations. Structural and biochemical results indicate that octamerization is required for substrate binding and catalysis by Sa_enolase.
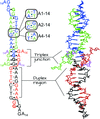
The X-ray crystal structure of a self-assembling DNA 14-mer containing a tertiary-structural mimic of tandem G–A base pairs is reported.
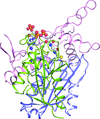
The crystal structures of a T1-like thiolase from M. smegmatis in three apo forms and one liganded form reveal an asymmetric tetrameric organization and a novel substrate-specificity pocket.
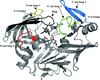
The crystal structure of the secreted aspartic protease Sapp2p from C. parapsilosis at an atomic resolution of 0.825 Å, the highest resolution aspartic protease structure deposited in the PDB to date, is reported.
PDB reference: Sapp2p, 4y9w
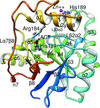
The first crystal structure of a U32 peptidase domain shows a TIM barrel-type architecture with an extended β7–β8 connection. There are no discernible motifs that are suitable for catalysing peptide-bond hydrolysis.
PDB reference: Mk0906, 5d88

Serial crystallography enables the extraction/purification of single-phase subsets from a mixture of multiphase diffraction snapshots.
Open access
access
Sulfur SAD phasing facilitates the structure determination of diverse native proteins using femtosecond X-rays from free-electron lasers via serial femtosecond crystallography.
PDB reference: lysozyme, 4yop
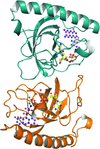
The FAD synthetase from C. ammoniagenes (CaFADS) is a bifunctional enzyme that folds into two almost independent modules. Binding of ligands to its individually expressed C-terminal riboflavin kinase (RFK) module triggers important structural changes. Their evaluation provides structural insights into the conformational changes that take place during the RFK catalytic cycle of CaFADS.







