journal menu
journal menu





issue contents
February 2016 issue
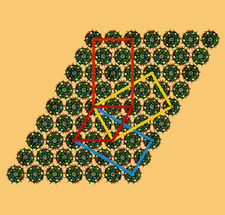
Cover illustration: A chimeric DNA-RNA Z-type duplex in complex with Ba2+ ions (Gilski et al., p. 211). Primitive monoclinic P cell (red) with a = c, ![[beta]](/logos/entities/beta_rmgif.gif) = 120° and the three C-centred (orthohexagonal) cells coloured blue (setting C1), yellow (setting C2) and orange (setting C3), shown together with the DNA-RNA-Ba2+ chimera packing viewed along b. The duplexes are packed in the ac plane and are located on the 21 axes. The origin is the same for all cells.
= 120° and the three C-centred (orthohexagonal) cells coloured blue (setting C1), yellow (setting C2) and orange (setting C3), shown together with the DNA-RNA-Ba2+ chimera packing viewed along b. The duplexes are packed in the ac plane and are located on the 21 axes. The origin is the same for all cells.
research papers






A protein complex from Y. entomophaga was crystallized and its structure was determined. Phasing this structure was a challenging problem owing to its large size (two heterodimers; a total of 4354 amino acids in the asymmetric unit) and its sensitivity to radiation damage. Low-resolution phase information from a Ta6Br12 derivative was transferred to a non-isomorphous selenomethionine-derivatized data set, allowing the heavy-atom substructure to be solved from an anomalous difference Fourier map.

The structural characterization of the N-terminal part of the nucleocapsid from Middle East respiratory syndrome coronavirus (MERS-CoV), a recently emerging virus, is reported. The structure of the N-terminal region, which includes a disordered tail followed by a globular domain, was obtained by combining X-ray diffraction and SAXS.
PDB reference: N-terminal domain of the MERS-CoV nucleocapsid, 4ud1

The three-dimensional structure of the C212S mutant of uridine phosphorylase from S. oneidensis MR-1 was determined at 1.68 Å resolution. Binding of the substrate was shown to require the concerted action of two subunits of the functional dimer.
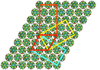
The self-complementary dCrGdCrGdCrG hexanucleotide with not only alternating pyrimidine/purine bases but also ribo/deoxy sugars crystallized in the presence of barium cations in the form of a left-handed Z-type duplex. While the structure has monoclinic symmetry, the crystals show complicated twinning, with the diffraction pattern arranged on a hexagonal lattice.
PDB reference: dCrGdCrGdCrG, 5ebi
Open  access
access
access
Observations are presented from retrospective analyses of the crystallization strategies deployed at the SGC, Oxford during its first decade of existence, providing practical guidelines for the design of screening experiments.

The crystal structure of BamB from E. coli in complex with the POTRA3–4 domains of BamA is reported.
PDB reference: complex of BamB with BamA POTRA3–4 domains, 4xga
The cap-binding domain of influenza virus protein PB2 is an excellent drug target, but there are some variations among different virus strains. This work provides insights into how an inhibitor may be designed to target multiple strains of both type A and type B influenza viruses.
Open access
access
The three-dimensional structures of two industrially important family GH3 β-D-glucosidases from A. fumigatus and A. oryzae are reported at 1.95 Å resolution. Both enzymes show extensive N-glycosylation. The extensive glycans pose special problems for crystallographic refinement, and new techniques and protocols were developed especially for this work.
Open access
access
A new method for automatic modeling of side-chain conformations that takes advantage of rotamer-prediction methods in a crystallographic context can improve the accuracy and validity of crystallographic models.
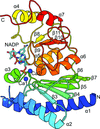
The NADP+-bound crystal structure of staphylococcal dual-specific IMPase/NADP(H) phosphatase (SaIMPase-I) represents a molecular glimpse of a substrate-bound structure of an NADP(H) phosphatase with the bound NADP+ demonstrating a folded conformation. The bound NADP+ in the active site of SaIMPase-I also represents a molecular scenario of protein–NADP+ interaction in which the hydride-transfer reaction of the nicotinamide ring is not the primary objective.