

 journal menu
journal menu




issue contents
September 2016 issue
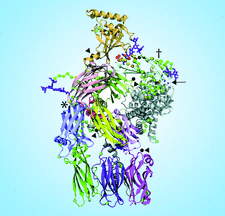
Cover illustration: Use of the recently developed interactive molecular-dynamics flexible fitting (iMDFF) approach significantly reduces the complexity of the conformational space to be searched during manual rebuilding of macromolecules. iMDFF has been used to individually correct and re-refine three structures and strategies for working with such challenging data sets are suggested (Croll & Andersen, p. 1006). Notably, the improved model allowed the resolution for complement C4b to be extended from 4.2 to 3.5 Å as demonstrated by paired refinement. This image gives an overview of the C4 structure and the key corrections made in this study. Register errors affecting two ![[beta]](/logos/entities/beta_rmgif.gif) -strands and a poorly ordered loop in the MG3 domain (marked with an asterisk) and four -strands in the CUB domain (marked with a dagger) were corrected with shifts of 1-3 residues. All C
-strands and a poorly ordered loop in the MG3 domain (marked with an asterisk) and four -strands in the CUB domain (marked with a dagger) were corrected with shifts of 1-3 residues. All C![[alpha]](/logos/entities/alpha_rmgif.gif) atoms which shifted by more than 2.5 Å are shown in sphere representation and coloured according to the distance moved. Additionally, four low-occupancy trimethyllead ions were resolved (black spheres and arrowheads) as well as the O-linked N-acetylgalactosamine (arrow; obscured behind protein in this image).
atoms which shifted by more than 2.5 Å are shown in sphere representation and coloured according to the distance moved. Additionally, four low-occupancy trimethyllead ions were resolved (black spheres and arrowheads) as well as the O-linked N-acetylgalactosamine (arrow; obscured behind protein in this image).
research papers








 access
access access
access access
access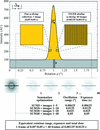
 access
access
addenda and errata
access
book reviews








