

 journal menu
journal menu





issue contents
October 2019 issue
Cover illustration: Recent developments in the Phenix software package for macromolecular structure determination using X-rays, neutrons and electrons [Liebschner et al. (2019), Acta Cryst. D75, 861-877].
feature articles




Open  access
access
access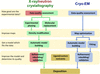
Recent developments in the Phenix software package are described in the context of macromolecular structure determination using X-rays, neutrons and electrons.
scientific commentaries
Open access
access
In structural biology, deriving and refining atomic models into maps obtained from X-ray crystallography or cryo electron microscopy (cryo-EM) is essential for the detailed interpretation of a structure and its functional implications through interactions so that for example hydrogen bonds, drug specificity and associated molecular mechanisms can be analysed. This commentary summarizes the latest features of the Phenix software and also highlights the fact that cryo-EM increasingly contributes to data depositions in the PDB and EMDB.
CCP-EM
Open access
access
Flexible workflows for on-the-fly electron-microscopy single-particle image processing using Scipion
The Scipion framework allows very flexible image-processing workflows to be generated and employed at electron-microscopy facilities, such that image acquisition can be monitored and possible problems detected, thereby enabling early decisions to be made on the fly. The streaming workflow can be very simple or extended, permitting the data resolution and heterogeneity to be estimated and adapted to the desire of the user and the microscope operator.
research papers


Open access
access
In this work, the X-ray crystal structures of four different deuterium-labelled versions of a surface variant of human carbonic anhydrase IX are compared and discussed. The results show that the overall structure and active-site organization of each version are essentially the same, paving the way for future neutron protein crystallography studies.
PDB references: human carbonic anhydrase IX, protiated, 6rqn; 6rqq; H/D-exchanged, 6rqu; deuterated, 6rqw

Mason–Pfizer monkey virus protease has been crystallized in a dimeric form using nearly the same conditions as those that yielded crystals of the monomeric form. A comparison of the structures of the two states highlights the conformational changes accompanying retropepsin dimerization.
Open access
access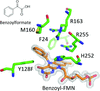
Structural and enzymological explorations of p-hydroxy-mandelate oxidase and its mutants uncover an unprecedented electrophilic/nucleophilic duality for the flavin mononucleotide cofactor as well as an intramolecular disproportionation mechanism for an oxidative decarboxylation reaction.
PDB references: p-hydroxymandelate oxidase, 5zzp; complex with (S)-mandelate, 5zzr; complex with benzoylformate, 6a08; Y128C mutant, complex with benzoylformate, 5zzz; Y128F mutant, 6a13; complex with (S)-mandelate, 6a0v; complex with 5-deazariboflavin mononucleotide, 6a1h; complex with 5-deazariboflavin mononucleotide and benzoic acid, 6a1l; complex with 5-deazariboflavin mononucleotide and benzoylformate, 6a1m; complex with 5-deazariboflavin mononucleotide and phenylpyruvate, 6a1p; complex with phenylpyruvate and riboflavin mononucleotide, 6a1r; complex with benzoylformate, 6a19; complex with malonyl–riboflavin mononucleotide, 6a21; complex with benzoylformate and riboflavin mononucleotide, 6a23; R163L mutant, complex with mandelamide–riboflavin mononucleotide, 6a3t

The structure of the human nucleosome core particle was solved by selenium SAD phasing. The present study demonstrates that nucleosome structures can be readily determined by experimental phasing and provides a method for solving nucleosome structures by X-ray crystallography in cases where the phase information is difficult to obtain by molecular replacement.
Open access
access
A novel method is presented to screen for suitable crystallization conditions and produce large amounts of microcrystals of membrane proteins in lipidic cubic phase for serial crystallography experiments.







