

 journal menu
journal menu





issue contents
March 2020 issue
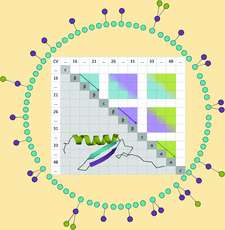
Cover illustration: Scheme of the fold-extraction algorithm in ALEPH, fragment-based molecular-replacement software which is optimized for the analysis and extraction of small protein folds by relying on their geometry rather than on their sequence [Medina et al. (2020), Acta Cryst. D76, 193-208].
CCP4




Open  access
access
access
ALEPH characterizes the main-chain geometry of small, noncontinuous fragments to flexibly annotate secondary structure, decompose folds, extract libraries and superpose fragments. Secondary and tertiary structure are described through networks of characteristic vectors, which are defined between the centroids of the Cα and carbonyl O atoms in a peptide.
Open access
access
The software ALIXE, which is available as a standalone program as well as integrated into the ARCIMBOLDO programs, combines partial solutions in reciprocal space to increase the signal and reduce the number of intermediate solutions. Its new implementation is faster and can be used by default in the process of phasing with fragments even on modest hardware.
Open access
access
When phasing cannot be accomplished from a partial polyalanine starting model, extending the model with side chains in a multi-solution way may succeed. SEQUENCE SLIDER implements this approach for use in ARCIMBOLDO.
Open access
access
The information content gained by making a diffraction-intensity measurement is a natural criterion for deciding which data make a useful contribution and which can legitimately be omitted from a calculation.

Open access
access
A novel method for the enhancement of automated protein model building using polypeptide fragments of homologous structures is presented.

Open access
access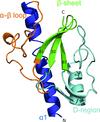
A practical perspective on molecular-replacement (MR) structure-determination pipelines is presented, using PilA1 from Clostridioides difficile as an example of a difficult case. A manual approach informed by the biology of the system under study is described, together with ab initio MR structure determination.
PDB reference: PilA1, 6t8s
Open access
access
The solution of coiled-coil crystal structures may be achieved by AMPLE through the use of ensembled ab initio models in molecular replacement. Improvements in ab initio modelling of elongated helices and oligomeric coiled-coils allow AMPLE to solve a greater number of coiled-coil structures and at lower resolution than previously achieved.
Open access
access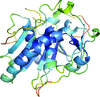
Local error estimates can be used to improve the success of models in molecular replacement.
research papers

Type 2 cysteine desulfurases are ubiquitous and often essential in plants and microorganisms owing to their role in Fe–S cluster biosynthesis. Here, an X-ray crystal structure of wild-type SufS with a rotated active-site β-hairpin, and a global structure and sequence analysis that argues for a conserved β-latch mechanism regulating type 2 cysteine desulfurases, are reported.
PDB reference: SufS with a spontaneously rotated β-hairpin, 6uy5

In the present study, it is shown that refinement and the resulting structures of hexagonal HIV-1 protease cocrystal structures can be markedly improved by choosing a lower symmetry space group (P61) and applying a twin law (h, −h − k, −l) to correct for merohedral twinning.







