Buy article online - an online subscription or single-article purchase is required to access this article.
The title compound, C
8H
5NO, has an intramolecular O

CN contact involving an O
C distance of 2.797 (2) Å and a C—C—N bond angle of 174.5 (2)°, both indicative of a weak nucleophilic attack of the aldehyde O atom on the electrophilic C atom in the nitrile group. Calculations at the B3LYP density functional level using the 6–31G* basis set support this interpretation; natural bond-order analysis indicates an
nO1 → π
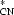
delocalization energy of 6.3 kJ mol
−1. Similar results were obtained from density functional calculations on three related molecules. The 2-formylbenzonitrile molecules pack in sheets as a consequence of C—H
N and C—H
O hydrogen bonds.
Supporting information
CCDC reference: 612446
The compound was obtained from Aldrich. The crystal used for diffraction was grown from chloroform.
H atoms were introduced at calculated positions, with C—H distances of 0.95 Å, and refined using a riding model [Uiso(H) = 1.2Ueq(C)].
Data collection: SMART (Bruker, 2002); cell refinement: SAINT (Bruker, 2002); data reduction: SAINT; program(s) used to solve structure: SHELXTL (Sheldrick, 1997); program(s) used to refine structure: SHELXLTL; molecular graphics: SHELXLTL; software used to prepare material for publication: SHELXLTL.
Crystal data top
| C8H5NO | F(000) = 272 |
| Mr = 131.13 | Dx = 1.374 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2ybc | Cell parameters from 1810 reflections |
| a = 3.7889 (9) Å | θ = 2.3–24.3° |
| b = 15.580 (4) Å | µ = 0.09 mm−1 |
| c = 10.759 (3) Å | T = 173 K |
| β = 93.73 (3)° | Needle, colorless |
| V = 633.8 (3) Å3 | 0.50 × 0.05 × 0.05 mm |
| Z = 4 | |
Data collection top
Siemens SMART area-detector
diffractometer | 974 reflections with I > 2σ(I) |
| Radiation source: fine-focus sealed tube | Rint = 0.041 |
| Graphite monochromator | θmax = 25.0°, θmin = 2.3° |
| ω scans | h = −4→4 |
| 3166 measured reflections | k = −18→15 |
| 1115 independent reflections | l = −12→12 |
Refinement top
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.042 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.101 | H-atom parameters constrained |
| S = 1.08 | w = 1/[σ2(Fo2) + (0.038P)2 + 0.26P]
where P = (Fo2 + 2Fc2)/3 |
| 1115 reflections | (Δ/σ)max = 0.001 |
| 91 parameters | Δρmax = 0.15 e Å−3 |
| 0 restraints | Δρmin = −0.17 e Å−3 |
Crystal data top
| C8H5NO | V = 633.8 (3) Å3 |
| Mr = 131.13 | Z = 4 |
| Monoclinic, P21/c | Mo Kα radiation |
| a = 3.7889 (9) Å | µ = 0.09 mm−1 |
| b = 15.580 (4) Å | T = 173 K |
| c = 10.759 (3) Å | 0.50 × 0.05 × 0.05 mm |
| β = 93.73 (3)° | |
Data collection top
Siemens SMART area-detector
diffractometer | 974 reflections with I > 2σ(I) |
| 3166 measured reflections | Rint = 0.041 |
| 1115 independent reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.042 | 0 restraints |
| wR(F2) = 0.101 | H-atom parameters constrained |
| S = 1.08 | Δρmax = 0.15 e Å−3 |
| 1115 reflections | Δρmin = −0.17 e Å−3 |
| 91 parameters | |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| O1 | 0.2865 (4) | 0.69998 (8) | 0.88147 (12) | 0.0395 (4) | |
| N1 | 0.4433 (4) | 0.87051 (10) | 1.05357 (14) | 0.0372 (4) | |
| C1 | 0.1212 (4) | 0.88277 (10) | 0.83388 (14) | 0.0251 (4) | |
| C2 | 0.0145 (4) | 0.81241 (10) | 0.75790 (15) | 0.0246 (4) | |
| C3 | −0.1618 (4) | 0.82789 (11) | 0.64310 (15) | 0.0275 (4) | |
| H3 | −0.2372 | 0.7809 | 0.5917 | 0.033* | |
| C4 | −0.2293 (4) | 0.91114 (11) | 0.60254 (15) | 0.0309 (4) | |
| H4 | −0.3470 | 0.9209 | 0.5231 | 0.037* | |
| C5 | −0.1256 (4) | 0.97997 (11) | 0.67752 (16) | 0.0323 (4) | |
| H5 | −0.1739 | 1.0369 | 0.6497 | 0.039* | |
| C6 | 0.0491 (4) | 0.96605 (11) | 0.79340 (15) | 0.0295 (4) | |
| H6 | 0.1192 | 1.0134 | 0.8449 | 0.035* | |
| C7 | 0.3021 (4) | 0.87205 (10) | 0.95581 (15) | 0.0280 (4) | |
| C8 | 0.0904 (4) | 0.72222 (11) | 0.79371 (15) | 0.0300 (4) | |
| H8 | −0.0231 | 0.6785 | 0.7443 | 0.036* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| O1 | 0.0500 (8) | 0.0320 (7) | 0.0352 (7) | 0.0035 (6) | −0.0076 (6) | 0.0019 (5) |
| N1 | 0.0423 (9) | 0.0381 (9) | 0.0304 (8) | 0.0009 (7) | −0.0047 (7) | −0.0030 (7) |
| C1 | 0.0221 (8) | 0.0288 (9) | 0.0242 (8) | −0.0002 (7) | 0.0007 (7) | 0.0001 (7) |
| C2 | 0.0232 (8) | 0.0256 (8) | 0.0254 (8) | −0.0008 (7) | 0.0042 (6) | −0.0008 (6) |
| C3 | 0.0269 (9) | 0.0314 (9) | 0.0242 (9) | −0.0025 (7) | 0.0005 (7) | −0.0042 (7) |
| C4 | 0.0296 (9) | 0.0373 (10) | 0.0254 (9) | 0.0012 (8) | −0.0018 (7) | 0.0043 (7) |
| C5 | 0.0336 (9) | 0.0274 (9) | 0.0356 (10) | 0.0018 (7) | −0.0010 (8) | 0.0064 (8) |
| C6 | 0.0291 (9) | 0.0272 (9) | 0.0318 (9) | −0.0015 (7) | −0.0003 (7) | −0.0041 (7) |
| C7 | 0.0288 (9) | 0.0260 (8) | 0.0294 (9) | −0.0003 (7) | 0.0030 (7) | −0.0021 (7) |
| C8 | 0.0333 (9) | 0.0294 (9) | 0.0274 (9) | −0.0018 (7) | 0.0024 (7) | −0.0031 (7) |
Geometric parameters (Å, º) top
| O1—C8 | 1.214 (2) | C3—H3 | 0.9500 |
| N1—C7 | 1.149 (2) | C4—C5 | 1.383 (2) |
| C1—C6 | 1.390 (2) | C4—H4 | 0.9500 |
| C1—C2 | 1.411 (2) | C5—C6 | 1.390 (2) |
| C1—C7 | 1.450 (2) | C5—H5 | 0.9500 |
| C2—C3 | 1.387 (2) | C6—H6 | 0.9500 |
| C2—C8 | 1.480 (2) | C8—H8 | 0.9500 |
| C3—C4 | 1.387 (2) | | |
| | | |
| C6—C1—C2 | 120.05 (14) | C3—C4—H4 | 119.9 |
| C6—C1—C7 | 117.57 (14) | C4—C5—C6 | 120.18 (16) |
| C2—C1—C7 | 122.38 (15) | C4—C5—H5 | 119.9 |
| C3—C2—C1 | 118.96 (15) | C6—C5—H5 | 119.9 |
| C3—C2—C8 | 118.10 (14) | C5—C6—C1 | 119.94 (15) |
| C1—C2—C8 | 122.92 (15) | C5—C6—H6 | 120.0 |
| C2—C3—C4 | 120.75 (15) | C1—C6—H6 | 120.0 |
| C2—C3—H3 | 119.6 | N1—C7—C1 | 174.54 (17) |
| C4—C3—H3 | 119.6 | O1—C8—C2 | 124.91 (16) |
| C5—C4—C3 | 120.12 (15) | O1—C8—H8 | 117.5 |
| C5—C4—H4 | 119.9 | C2—C8—H8 | 117.5 |
| | | |
| C6—C1—C2—C3 | 0.0 (2) | C3—C4—C5—C6 | 0.5 (3) |
| C7—C1—C2—C3 | 179.10 (15) | C4—C5—C6—C1 | 0.3 (2) |
| C6—C1—C2—C8 | 178.42 (15) | C2—C1—C6—C5 | −0.5 (2) |
| C7—C1—C2—C8 | −2.5 (2) | C7—C1—C6—C5 | −179.65 (15) |
| C1—C2—C3—C4 | 0.7 (2) | C3—C2—C8—O1 | 168.30 (16) |
| C8—C2—C3—C4 | −177.73 (15) | C1—C2—C8—O1 | −10.1 (3) |
| C2—C3—C4—C5 | −1.0 (2) | | |
Experimental details
| Crystal data |
| Chemical formula | C8H5NO |
| Mr | 131.13 |
| Crystal system, space group | Monoclinic, P21/c |
| Temperature (K) | 173 |
| a, b, c (Å) | 3.7889 (9), 15.580 (4), 10.759 (3) |
| β (°) | 93.73 (3) |
| V (Å3) | 633.8 (3) |
| Z | 4 |
| Radiation type | Mo Kα |
| µ (mm−1) | 0.09 |
| Crystal size (mm) | 0.50 × 0.05 × 0.05 |
| |
| Data collection |
| Diffractometer | Siemens SMART area-detector
diffractometer |
| Absorption correction | – |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 3166, 1115, 974 |
| Rint | 0.041 |
| (sin θ/λ)max (Å−1) | 0.595 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.042, 0.101, 1.08 |
| No. of reflections | 1115 |
| No. of parameters | 91 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.15, −0.17 |
Distancesa and angles (Å, °) in the C—H···X—C contacts top| C—H···X—C | C—H···X | H···X | H···X—C | C···X |
| C3—H3···N1i—C7i | 152 | 2.67 | 110 | 3.540 (2) |
| C5—H5···O1ii—C8ii | 169 | 2.60 | 95 | 3.532 (2) |
| C6—H6···N1iii—C7iii | 157 | 2.64 | 127 | 3.533 (2) |
| Note: (a) all C—H distances are 0.95 Å. Symmetry codes: (i) −1 + x, 1/2 − y, −1/2 + z; (ii) 1 − x, 1/2 + y, 3/2 − z; (iii) 2 − x, 1 − y, 2 − z. |
Experimental and computed values for substituted aryl nitriles (Å, °) top | (I)a | (II)b | (III)c | (IV)d |
| Experimental | | | | |
| O···C | 2.797 (2) | 2.837 (4) | 2.607 (3) | 2.758 (4) |
| C—C—N | 174.5 (2) | 176.8 (4) | 174.0 (2) | 174.4 (5) |
| C—C—O | 124.9 (2) | 119.9 (3) | 118.8 (2) | 123.4 (4) |
| O—C—C—C | 10.1 (3) | 42.6 (4) | 12.1 (3) | 7.4 (5) |
| | | | |
| Computed | | | | |
| O···C | 2.806 | 2.858 | 2.665 | 2.758 |
| C—C—N | 173.0 | 174.7 | 171.4 | 172.9 |
| C—C—O | 125.3 | 119.0 | 120.2 | 123.6 |
| O—C—C—C | 0.0 | 39.7 | 17.0 | 6.0 |
| | | | |
| O···C bond order | 0.022 | 0.005 | 0.023 | 0.023 |
| C—O bond order | 1.908 | 1.829 | 1.811 | 1.841 |
| C—N bond order | 2.765 | 2.784 | 2.759 | 2.766 |
| NBO nO1 → σ*CN kJ mol−1 | 6.3 | 2.5 | 9.6 | 7.9 |
| Notes: (a) o-nitrobenzaldehyde; (b) 2-cyanobenzophenone; (c) 9-oxo-1-thioxanthenecarbonitrile; (d) 5-cyano-7-methoxy-2,2-dimethyl-4-oxo-chroman-6-yl acetate; (e) torsion angle of the CCO group with respect to the mean plane of the C6 ring. |

 Subscribe to Acta Crystallographica Section C: Structural Chemistry
Subscribe to Acta Crystallographica Section C: Structural Chemistry
The full text of this article is available to subscribers to the journal.
If you have already registered and are using a computer listed in your registration details, please email
[email protected] for assistance.


 journal menu
journal menu organic compounds
organic compounds










![[Figure 1]](https://journals.iucr.org/c/issues/2006/06/00/jz3013/jz3013fig1thm.gif)
![[Figure 2]](https://journals.iucr.org/c/issues/2006/06/00/jz3013/jz3013fig2thm.gif)
![[Figure 3]](https://journals.iucr.org/c/issues/2006/06/00/jz3013/jz3013fig3thm.gif)




In three o-nitrobenzonitrile derivatives (5-chloro-, 6-chloro- and 6-methyl-) there are weak electrophilic nucleophilic (or Lewis acid–base) interactions between a nitro O atom and the nitrile C atom (Britton & Cramer, 1996). This situation is shown by a short O···C distance and a bending of the C—CN group away from the interacting O atom. The study of o-cyanobenzaldehyde reported here was undertaken to see whether the same (expected) interaction occurred.
The atoms labelling and displacement ellipsoids are shown in Fig. 1, which also shows the O···C interaction. The O1···C7 distance is 2.797 (2) Å, which can be compared with the van der Waals distance of 3.22 (Bondi, 1964; Rowland & Taylor, 1996). The C1—C7—N1 angle is 174.5 (2)° and the CN group bends away from atom O1. This is similar to the results in the o-nitrobenzonitriles except that the O···C distance is about 0.2 Å longer in the aldehyde. This difference is reasonable since the aldehyde O atom has no formal change, while the nitro O atom has a formal change of −1/2. The other bond angles and distances in the molecule are normal.
The molecules pack in sheets parallel to (102) (Fig. 2). The molecules are tilted by 7.8 (1)° with respect to the sheets. There are three weak C—H···X interactions with H···X distances less than 2.70 Å. These interactions give rise to three different rings as can be seen in Fig. 2. Using graph-set notation (Etter, 1990; Bernstein et al., 1995) there is an R22(10) ring involving two H6 atoms, an R33(10) ring involving atoms H3, H6 and H5, and an R44(22) ring involving two H3 and two H5 atoms. The geometric data for these interactions are given in Table 1.
The Cambridge Structure Database (Allen, 2002) contains three additional structures with short O···CN interactions, viz. 2-cyanobenzophenone, (II) (Preut et al., 1992), 9-oxo-1-thioxanthenecarbonitrile, (III) (Abboud et al., 1990), and 5-cyano-7-methoxy,2,2-dimethyl-4-oxo-chroman-6-yl acetate, (IV) (Clegg, 2003). These are shown in Fig. 3. Relevant experimental data for these compounds are given in Table 2, where they are compared with electronic structure calculations.
To provide further insight into interactions between the potentially interacting carbonyl and nitrile groups in (I)–(IV), electronic structure calculations were carried out at the density functional level (Cramer, 2004) using the 6–31G* basis set (Hehre et al., 1986) and the hybrid B3LYP functional (Becke, 1988; Lee et al., 1988; Becke, 1993; Stephens et al., 1994). All structures were fully optimized without the imposition of any symmetry constraints. The optimized densities were employed in the computation of interatomic bond orders according to the scheme of Mayer (1983). In addition, the energetic stabilization associated with the nO1 → π*CN interaction was quantified by second-order perturbation theory in the natural bond orbital (NBO) basis (Reed et al., 1988). CM3 partial atomic charges (Winget et al., 2002) were also examined but charges on the nitrile N atoms in (I)–(IV) varied negligibly.
Although the calculated gas-phase and experimental solid-state structures should not be expected to match completely, key trends in the two sets of structures are manifest. Most noteworthy is the increasing deviation of the nitrile N atom from collinearity with the nitrile C atom and the ipso C atom of the aromatic ring as the distance between the carbonyl O and the nitrile C atoms decreases. If the carbon–oxygen interaction were purely repulsive, the nitrile group would be expected to be displaced away from the O atom, but that does not require bending at the nitrile C atom.
The presence of substantial repulsion is certainly supported by other geometric parameters. For instance, as the OCCC dihedral angle is reduced, the CCO angle increases to maintain separation from the nitrile C atom. In the case of o-cyanobenzaldehyde, where conjugation evidently favors coplanarity of all of the exocyclic functionality, the CCO bond angle increases to 125.3°.
Analysis of the B3LYP densities provides in each case some support for an incipient nucleophilic interaction between the carbonyl O atom and nitrile C atom. That interaction is not sufficient to lead to any significant Mayer bond order developing between the two atoms. Instead, NBO analysis suggests that there is a stabilization associated with an nO1 → π*CN interaction that increases steadily with decreasing O—C distance and concomitant decreasing C—C—N angles. The bending of the nitrile group facilitates this delocalization by increasing the amplitude of the π* orbital on the side of the bond adjacent to the carbonyl O atom. This effect does lead to a small reduction in the C—N Mayer bond order as the bond angle increasingly deviates from linearity.