Buy article online - an online subscription or single-article purchase is required to access this article.
The title compound, [Ni(NCS)
2(C
5H
4N
4O)
2(H
2O)
2], crystallizes in the triclinic space group
P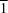
. The molecular unit contains two neutral molecules of 4,5-dihydro-1,2,4-triazolo[1,5-
a]pyrimidin-5-one (5HtpO) coordinated through the N atom in position 3, two thiocyanate ligands coordinated through their N atoms and two water molecules completing an octahedral environment around the Ni
II ion, which lies on a centre of inversion. The structure is stabilized by hydrogen bonding. Distances in the coordination sphere are Ni-N3(5HtpO) 2.132 (2), Ni-O(water) 2.085 (2) and Ni-N(thiocyanato) 2.040 (2) Å.
Supporting information
CCDC reference: 150312
The ligand 5HtpO was prepared according to the published method (Abul Haj et al., 2000) from 3-amino-[1,2,4]-triazole and ethyl 3,3-diethoxypropionate. The nickel complex was prepared by mixing three solutions, 10 ml each, the first one containing 0.11 g of nickel nitrate hexahydrate in water, the second with 0.075 g KSCN in water, and the third with 0.136 g of the ligand 5HtpO in acetonitrile:water (2:1). The resulting solution was filtered off and left to evaporate at room temperature. Green crystals of the compound were obtained after 5 d, collected by filtration and air dried.
H atoms attached to C and N were introduced in their ideal positions and allowed to ride on their parent atoms. Those of the water ligand were located in the difference maps and refined while restraining the O—H distance to 0.84 Å. The displacement parameters of the H atoms were constrained to 1.2Ueq of their parent atoms.
Data collection: STADI4 (Stoe & Cie, 1996); cell refinement: STADI4; data reduction: X-RED (Stoe & Cie, 1996); program(s) used to solve structure: SHELXS86 (Sheldrick, 1990); program(s) used to refine structure: SHELXL93 (Sheldrick, 1993); molecular graphics: Xtal_GX (Hall & du Boulay, 1997).
Crystal data top
| [Ni(CNS)2(C5H4N4O)2(H2O)2] | Z = 1 |
| Mr = 483.15 | F(000) = 246 |
| Triclinic, P1 | Dx = 1.703 Mg m−3 |
| a = 7.8040 (3) Å | Mo Kα radiation, λ = 0.71073 Å |
| b = 7.9946 (3) Å | Cell parameters from 54 reflections |
| c = 8.9870 (3) Å | θ = 12.5–18.7° |
| α = 106.331 (3)° | µ = 1.30 mm−1 |
| β = 108.534 (3)° | T = 293 K |
| γ = 105.041 (3)° | Irregularly shaped crystals, green |
| V = 471.17 (3) Å3 | 0.50 × 0.35 × 0.18 mm |
Data collection top
STOE STADI4 4-circle-
diffractometer | 2390 reflections with I > 2σ(I) |
| Radiation source: fine-focus sealed tube | Rint = 0.000 |
| Graphite monochromator | θmax = 30.0°, θmin = 2.6° |
| Scan width (ω) = 1.05 – 1.25, scan ratio 2θ:ω = 1.00 | h = −10→10 |
Absorption correction: ψ scan
(X-RED; Stoe & Cie, 1996) | k = −11→10 |
| Tmin = 0.562, Tmax = 0.686 | l = 0→12 |
| 2746 measured reflections | 3 standard reflections every 60 min |
| 2746 independent reflections | intensity decay: 6.5% |
Refinement top
| Refinement on F2 | Primary atom site location: heavy-atom method |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.038 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.106 | H-atom parameters constrained |
| S = 1.26 | Weighting scheme based on measured s.u.'s w = 1/[σ2(Fo2) + (0.04P)2 + 0.25P]
where P = (Fo2 + 2Fc2)/3 |
| 2742 reflections | (Δ/σ)max = 0.001 |
| 139 parameters | Δρmax = 0.69 e Å−3 |
| 2 restraints | Δρmin = −0.37 e Å−3 |
Crystal data top
| [Ni(CNS)2(C5H4N4O)2(H2O)2] | γ = 105.041 (3)° |
| Mr = 483.15 | V = 471.17 (3) Å3 |
| Triclinic, P1 | Z = 1 |
| a = 7.8040 (3) Å | Mo Kα radiation |
| b = 7.9946 (3) Å | µ = 1.30 mm−1 |
| c = 8.9870 (3) Å | T = 293 K |
| α = 106.331 (3)° | 0.50 × 0.35 × 0.18 mm |
| β = 108.534 (3)° | |
Data collection top
STOE STADI4 4-circle-
diffractometer | 2390 reflections with I > 2σ(I) |
Absorption correction: ψ scan
(X-RED; Stoe & Cie, 1996) | Rint = 0.000 |
| Tmin = 0.562, Tmax = 0.686 | 3 standard reflections every 60 min |
| 2746 measured reflections | intensity decay: 6.5% |
| 2746 independent reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.038 | 2 restraints |
| wR(F2) = 0.106 | H-atom parameters constrained |
| S = 1.26 | Δρmax = 0.69 e Å−3 |
| 2742 reflections | Δρmin = −0.37 e Å−3 |
| 139 parameters | |
Special details top
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
Refinement. Refinement on F2 for ALL reflections except for 4 with very negative F2 or flagged by the user for potential systematic errors. Weighted R-factors wR and all goodnesses of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The observed criterion of F2 > σ(F2) is used only for calculating R_factor_gt etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| Ni | 0.5000 | 0.5000 | 0.5000 | 0.02740 (12) | |
| N1 | 0.4416 (3) | 0.6935 (3) | 0.0976 (3) | 0.0450 (5) | |
| C2 | 0.5232 (4) | 0.6802 (4) | 0.2429 (3) | 0.0403 (5) | |
| H2 | 0.6381 | 0.7747 | 0.3315 | 0.048* | |
| N3 | 0.4293 (3) | 0.5181 (3) | 0.2570 (2) | 0.0324 (4) | |
| C3A | 0.2780 (3) | 0.4241 (3) | 0.1059 (3) | 0.0290 (4) | |
| N4 | 0.1304 (3) | 0.2544 (3) | 0.0414 (2) | 0.0318 (4) | |
| H4 | 0.1310 | 0.1885 | 0.1023 | 0.038* | |
| C5 | −0.0215 (3) | 0.1831 (3) | −0.1191 (3) | 0.0325 (5) | |
| O5 | −0.1530 (3) | 0.0282 (2) | −0.1704 (2) | 0.0399 (4) | |
| C6 | −0.0124 (4) | 0.3009 (4) | −0.2158 (3) | 0.0414 (6) | |
| H6 | −0.1124 | 0.2603 | −0.3237 | 0.050* | |
| C7 | 0.1365 (4) | 0.4660 (4) | −0.1531 (3) | 0.0425 (6) | |
| H7 | 0.1431 | 0.5390 | −0.2171 | 0.051* | |
| N8 | 0.2800 (3) | 0.5256 (3) | 0.0084 (3) | 0.0347 (4) | |
| S | −0.17814 (9) | 0.12212 (9) | 0.28346 (10) | 0.0462 (2) | |
| C1T | 0.0484 (3) | 0.2736 (3) | 0.3815 (3) | 0.0306 (4) | |
| N1T | 0.2102 (3) | 0.3784 (3) | 0.4428 (3) | 0.0364 (4) | |
| O1W | 0.4902 (3) | 0.7667 (2) | 0.5865 (2) | 0.0394 (4) | |
| H11W | 0.399 (4) | 0.777 (5) | 0.615 (4) | 0.047* | |
| H12W | 0.596 (3) | 0.858 (4) | 0.657 (3) | 0.047* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| Ni | 0.0207 (2) | 0.0252 (2) | 0.0236 (2) | 0.00113 (14) | 0.00454 (14) | 0.00489 (14) |
| N1 | 0.0430 (12) | 0.0359 (11) | 0.0463 (12) | 0.0022 (9) | 0.0137 (10) | 0.0204 (10) |
| C2 | 0.0358 (12) | 0.0315 (11) | 0.0388 (12) | −0.0003 (9) | 0.0097 (10) | 0.0126 (10) |
| N3 | 0.0290 (9) | 0.0289 (9) | 0.0265 (9) | 0.0012 (7) | 0.0063 (7) | 0.0087 (7) |
| C3A | 0.0290 (10) | 0.0272 (10) | 0.0259 (9) | 0.0069 (8) | 0.0093 (8) | 0.0102 (8) |
| N4 | 0.0326 (9) | 0.0267 (9) | 0.0242 (8) | 0.0039 (7) | 0.0045 (7) | 0.0094 (7) |
| C5 | 0.0297 (10) | 0.0317 (11) | 0.0256 (10) | 0.0095 (9) | 0.0057 (8) | 0.0059 (8) |
| O5 | 0.0328 (9) | 0.0314 (8) | 0.0339 (8) | 0.0035 (7) | 0.0016 (7) | 0.0057 (7) |
| C6 | 0.0418 (13) | 0.0445 (14) | 0.0285 (11) | 0.0138 (11) | 0.0053 (10) | 0.0148 (10) |
| C7 | 0.0474 (14) | 0.0460 (14) | 0.0349 (12) | 0.0173 (12) | 0.0118 (11) | 0.0240 (11) |
| N8 | 0.0360 (10) | 0.0314 (10) | 0.0323 (10) | 0.0073 (8) | 0.0112 (8) | 0.0156 (8) |
| S | 0.0265 (3) | 0.0352 (3) | 0.0590 (4) | 0.0007 (2) | 0.0162 (3) | 0.0067 (3) |
| C1T | 0.0287 (10) | 0.0305 (10) | 0.0299 (10) | 0.0087 (8) | 0.0118 (8) | 0.0114 (8) |
| N1T | 0.0276 (9) | 0.0355 (10) | 0.0340 (10) | 0.0039 (8) | 0.0090 (8) | 0.0090 (8) |
| O1W | 0.0290 (8) | 0.0295 (8) | 0.0419 (9) | 0.0044 (7) | 0.0095 (7) | 0.0023 (7) |
Geometric parameters (Å, º) top
| Ni—N1T | 2.040 (2) | C3A—N8 | 1.352 (3) |
| Ni—O1W | 2.085 (2) | N4—C5 | 1.382 (3) |
| Ni—N3 | 2.132 (2) | C5—O5 | 1.232 (3) |
| N1—C2 | 1.306 (3) | C5—C6 | 1.453 (3) |
| N1—N8 | 1.381 (3) | C6—C7 | 1.334 (4) |
| C2—N3 | 1.376 (3) | C7—N8 | 1.374 (3) |
| N3—C3A | 1.322 (3) | S—C1T | 1.633 (2) |
| C3A—N4 | 1.351 (3) | C1T—N1T | 1.161 (3) |
| | | |
| N1T—Ni—O1W | 90.01 (8) | C3A—N4—C5 | 122.3 (2) |
| N1T—Ni—N3 | 90.80 (8) | O5—C5—N4 | 119.3 (2) |
| O1W—Ni—N3 | 85.85 (8) | O5—C5—C6 | 124.7 (2) |
| C2—N1—N8 | 101.7 (2) | N4—C5—C6 | 116.0 (2) |
| N1—C2—N3 | 116.0 (2) | C7—C6—C5 | 121.4 (2) |
| C3A—N3—C2 | 102.4 (2) | C6—C7—N8 | 118.3 (2) |
| C3A—N3—Ni | 134.6 (2) | C3A—N8—C7 | 123.1 (2) |
| C2—N3—Ni | 120.9 (2) | C3A—N8—N1 | 109.7 (2) |
| N3—C3A—N4 | 130.9 (2) | C7—N8—N1 | 127.2 (2) |
| N3—C3A—N8 | 110.2 (2) | N1T—C1T—S | 176.3 (2) |
| N4—C3A—N8 | 118.9 (2) | C1T—N1T—Ni | 163.2 (2) |
Experimental details
| Crystal data |
| Chemical formula | [Ni(CNS)2(C5H4N4O)2(H2O)2] |
| Mr | 483.15 |
| Crystal system, space group | Triclinic, P1 |
| Temperature (K) | 293 |
| a, b, c (Å) | 7.8040 (3), 7.9946 (3), 8.9870 (3) |
| α, β, γ (°) | 106.331 (3), 108.534 (3), 105.041 (3) |
| V (Å3) | 471.17 (3) |
| Z | 1 |
| Radiation type | Mo Kα |
| µ (mm−1) | 1.30 |
| Crystal size (mm) | 0.50 × 0.35 × 0.18 |
| |
| Data collection |
| Diffractometer | STOE STADI4 4-circle-
diffractometer |
| Absorption correction | ψ scan
(X-RED; Stoe & Cie, 1996) |
| Tmin, Tmax | 0.562, 0.686 |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 2746, 2746, 2390 |
| Rint | 0.000 |
| (sin θ/λ)max (Å−1) | 0.703 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.038, 0.106, 1.26 |
| No. of reflections | 2742 |
| No. of parameters | 139 |
| No. of restraints | 2 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.69, −0.37 |
Selected geometric parameters (Å, º) top| Ni—N1T | 2.040 (2) | S—C1T | 1.633 (2) |
| Ni—O1W | 2.085 (2) | C1T—N1T | 1.161 (3) |
| Ni—N3 | 2.132 (2) | | |
| | | |
| N1T—Ni—O1W | 90.01 (8) | C2—N3—Ni | 120.9 (2) |
| N1T—Ni—N3 | 90.80 (8) | N1T—C1T—S | 176.3 (2) |
| O1W—Ni—N3 | 85.85 (8) | C1T—N1T—Ni | 163.2 (2) |
| C3A—N3—Ni | 134.6 (2) | | |

 Subscribe to Acta Crystallographica Section C: Structural Chemistry
Subscribe to Acta Crystallographica Section C: Structural Chemistry
The full text of this article is available to subscribers to the journal.
If you have already registered and are using a computer listed in your registration details, please email
[email protected] for assistance.


 journal menu
journal menu metal-organic compounds
metal-organic compounds











![[Figure 1]](https://journals.iucr.org/c/issues/2000/08/00/ln1106/ln1106fig1thm.gif)




1,2,4-Triazolo[1,5-a]pyrimidine derivatives are interesting compounds for studying their interaction with metal ions since their molecules present several possible binding sites. This explains their versatile coordination chemistry, which has recently been reviewed by our group (Salas et al., 1999). This versatile behaviour is enhanced by the presence of different auxiliary ligands, thiocyanate being one such ligand that leads to heterometallic compounds (Biagini-Cingi et al., 1985) and can act as a very efficient transmitter of magnetic interaction (Navarro et al., 1997). Following this line, we have studied the interaction of NiII with the ligand 4,5-dihydro-5-oxo-[1,2,4]triazolo-[1,5-a]pyrimidine (5HtpO) in the presence of thiocyanate, a simple mononuclear compound with formula [Ni(5HtpO)2(NCS)2(H2O)2], (I), having been obtained, the structure of which is described here. In this case, as expected from its monomeric nature, magnetic interaction among the metal centres is not observed and the compound shows a temperature-independent magnetic moment of 3.28 BM. \sch
The crystal structure is properly described in the centrosymmetric space group P1 with the asymmetric unit comprising half of the complex with the metal atom lying in a crystallographic inversion centre. The environment of this atom is octahedral with two 5HtpO ligands coordinated monodentately through the N atom in position three, which is the most frequent binding site for this type of ligands (Salas et al., 1999). Thiocyanate ions are linked to the metal though their N atoms and two water molecules complete the coordination sphere in an `all-trans' disposition. A view of the molecular structure of the compound is shown in Figure 1 indicating also the labelling scheme.
The Ni—N3 bond is somewhat longer than in nickel(II) complexes of the analogous ligand 5,7-dimethyl-[1,2,4]triazolo-[1,5-a]pyrimidine (2.07–2.09 Å; Lenstra et al., 1989), which may be attributed to a higher basicity of this latter ligand when compared with 5HtpO due to the absence of the exocyclic O atom. Coordination does not appreciably affect the geometry of the ligand, the difference in bond distances and bond angles compared with free 5HtpO (Abul Haj et al., 2000) being below 0.02 Å and 1°, respectively. The ligand is in its neutral form, so the H atom at position 4 keeps in place, which is confirmed by the formation of a hydrogen bond with the carbonyl group of a neighbouring molecule [N4···O5(-x, −y, −z), 2.834 (3) Å]. The H atoms of the water ligands are likewise involved in hydrogen bonding [O1W···O5(1 + x, 1 + y, 1 + z) and O1W···S(-x, 1 − y, 1 − z), distances 2.728 (2) and 3.231 (2) Å]. The contacts N4···O5(-x, −y, −z) and O1W···S(-x, 1 − y, 1 − z) join pairs of molecules related by an inversion centre whereas the contact O1W···O5(1 + x, 1 + y, 1 + z) creates chains in the [111] direction. The combination of the three types of hydrogen bonds gives rise to two-dimensional arrays which lie in the (011) plane.
The angle Ni—N1T—C1T deviates appreciably from linearity. Such a situation is not unusual for nickel-isothiocyanato complexes (Moore & Squattrito, 1999; Kersting et al., 1999) and the analogous compounds of other first transition metals (Biagini-Cingi et al., 1986); values can be found even below 140° (Battaglia et al., 1980). Conversely, examples with almost linear geometry have also been described (Fontaine et al., 1987), which shows the great flexibility of the metal-thiocyanato bond. The value of the M—N—C angle probably depends on the interaction with neighbouring species more than on the electronic structure of the complex itself.