
 journal menu
journal menu



issue contents
 | STRUCTURAL BIOLOGY |
ISSN: 2059-7983
CCP4 Study Weekend 2021
Integrating Structural Biology
Edited by Atlanta Cook, Dan Rigden and Anastassis Perrakis
This virtual issue contains articles from the 2021 CCP4 Study Weekend.





Open  access
access
access
Bioinformatics tools, primarily those available through the MPI Bioinformatics Toolkit, for the annotation of protein sequences are described. These include tools for the identification of homologs of known structure, protein domains, sequence repeats, coiled coils, transmembrane segments and signal sequences.
Open access
access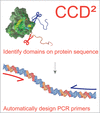
CCD2 is a software tool that aggregates sequence information for protein sequences (conservation, structure prediction, domain and disorder detection), enabling informed choices for expression-construct design, the single-click generation of PCR primers for cloning and easy data tracking.

Open access
access
The implications of the AlphaFold2 protein structure-modelling software for crystallographic phasing strategies are discussed.
Open access
access
MrParse is a software package designed to aid decision making in molecular replacement (MR). It performs a sequence search to find search models, not only in the PDB, as would conventionally be performed, but also in the EBI AlphaFold database, and provides a series of analyses relevant to MR such as crystal pathology detection and sequence analysis.
Open access
access
Ten goals for the future development of macromolecular refinement programs are described.
Open access
access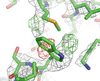
A new program, Servalcat, to facilitate atomic model refinement in cryo-EM single-particle analysis is presented. It implements a refinement pipeline using REFMAC5 and Fo − Fc map calculation.
Open access
access
Neutron protein crystallography provides insight into the structure and reaction mechanism of transition-state metal oxidoreductases without resulting in radiation-damage-induced artefacts.
Open access
access
Time-resolved cryo-EM allows the study of proteins under non-equilibrium conditions on the millisecond timescale. Here, in-flow and on-grid mixing techniques are directly compared and it is found that on-grid reactions can be influenced by air–water interactions, whilst in-flow reactions give a broader distribution of reaction times due to laminar flow.
Open access
access
The types, scopes, availability and complementarity of databases for intrinsically disordered proteins are described.
Open access
access
A brief overview of techniques used in the hit-identification/validation stage of drug discovery is given, highlighting the importance of orthogonal measurements and triangulation in early-stage crystallographic hit validation.
Open access
access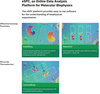
eSPC is an online tool to analyze biophysical data from fluorescence, microscale thermophoresis and differential scanning fluorimetry experiments. The modules from the data-analysis platform contain classical thermodynamic models and clear user guidelines for the determination of dissociation constants (Kd) and thermal unfolding parameters such as melting temperatures (Tm).
Open access
access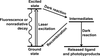
This review summarizes the best characterized and most relevant photocaging groups for time-resolved structural biology described in the literature to date. It provides a walkthrough of the essential factors to consider in designing a suitable photocaged molecule to address specific biological questions using time-resolved X-ray diffraction or solution-scattering methods.
Open access
access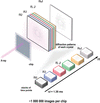
The key factors that should be considered during the planning and execution of a time-resolved crystallographic experiment are discussed, with a focus on time-resolved serial synchrotron crystallography.
Open access
access
A standalone system, called PDB-Dev, has been developed for archiving integrative structures and making them publicly available. The paper describes the data standards, the software tools and the various components of the PDB-Dev data-collection, processing and archiving infrastructure.
Open access
access
A 3D cellular imaging platform developed at beamline B24 at Diamond Light Source has been used to identify cellular features such as filamentous actin in mammalian cells. This approach enabled virtual same-sample imaging of structures captured on separate microscopes through rigid transformation of 3D data in silico, bypassing the need for additional sample processing and ensuring artefact-free data correlation.
Open access
access
Intracellular protein–ligand dissociation constants are measured by in-cell NMR spectroscopy by means of competition binding experiments with respect to a reference ligand. The method is applied to a set of carbonic anhydrase inhibitors, revealing intracellular binding with nanomolar affinity.
Open access
access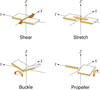
New restraints and validation targets for Watson–Crick base pairs are implemented in PDB-REDO and are applied to nucleic acid structures in the Protein Data Bank.
Open access
access
Utilities for atomic model validation in the CCP-EM software suite are discussed. An assessment of SARS-CoV-2 structures reflects a strong bias in model refinement towards geometric restraints when compared with agreement with the data.
Open access
access
An overview of computational docking and mapping approaches, and their biological applications to the problems of drug discovery, is presented.
Open access
access
The study of cells replicating hepatitis C and treated with antiviral compounds by soft X-ray cryo tomography (cryo-SXT) and synchrotron-based infrared microscopy allows correlation of the viral structures resolved by the former with their chemical composition revealed by the latter. The results show the potential of cryo-SXT as a platform to determine the effectiveness of antiviral compounds in infected cells, guiding drug development at a preclinical level.
Open access
access
Improvements to the ensemble refinement method are described and demonstrated. These improvements lead to more physically meaningful and interpretable macromolecular ensembles.







