journal menu
journal menu




issue contents
February 1999 issue
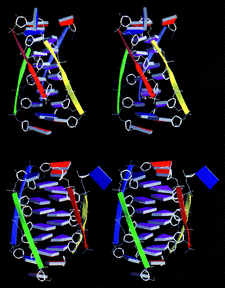
Cover illustration: Schematic diagram of the four-stranded d(ACCCT) molecule in two different orientations, showing the minor groove (top) and the major groove (bottom) (p. 422).
research papers




The determination of the structure of CysB(88–324) proved to be unusually difficult because of non-isomorphism, poor initial phase information resulting from weak and correlated heavy-atom substitution and missing low-resolution reflections. The strength of maximum-likelihood refinement (REFMAC) was employed to create a partial model which could eventually be used in multi-crystal averaging to solve the structure.

Download citation




Download citation
The structure of Ru(2,2′-bipyridine)2(imidazole) (His83)azurin (RuAz) has been determined to 2.3 Å resolution by X-ray crystallography. The structure of RuAz confirms that His83 is the only site of chemical modification and that the native azurin structure is not perturbed significantly by the ruthenium label.
PDB reference: ruthenium-modified azurin, 1bex
The X-ray crystal structure of free barnase has been refined anisotropically at 1.5 Å resolution and compared with the structures of barnase complexed with nucleotide inhibitors or barstar. The structural and functional role of water molecules is discussed.
PDB reference: barnase, 1a2p

Galactofuranose is a key component of the mycobacterial cell wall and thus a potential therapeutic target. The purification, crystallization and preliminary data collection of UDP-galactopyranose mutase from E. coli, the enzyme responsible for the conversion of UDP-galactopyranose into UDP-galactofuranose, is reported.
The crystal structure of recombinant human lactoferrin, overexpressed in a fungal system, shows that the structure of the protein is not affected by its different glycosylation, the strain improvement methods used, or the single mutation incorporated.
PDB reference: recombinant human lactoferrin, 1b0l

The structures of two crystal forms of the rubredoxin from D. vulgaris Miyazaki F have been solved by the molecular-replacement method. From the comparison of the crystal structure of the form II crystal with those of other rubredoxin molecules, a similarity in the core region, which is composed of aromatic residues, including the active center, has been revealed.
The structure analysis of P. laminosum plastocyanin at 2.8 Å resolution confirms that the polypeptide fold of cyanobacterial plastocyanins differs slightly but significantly from that of plant plastocyanins. The concentration of conserved surface negative charges found at the `acidic patch' of plant plastocyanins is absent.
PDB reference: plastocyanin, 1baw
The crystal structure of d(ACCCT) shows a four-stranded i-motif conformation. Noticeably, this complex is further stabilized by a three-base hydrogen-bonding network, in which two adenines and a thymine form four hydrogen bonds via a reverse Hoogsteen and an asymmetric adenine–adenine base pairing.
PDB reference: d(ACCCT), 1bqj

The replacement of all tryptophan residues by aza-tryptophans in the bacteriophage λ lysozyme was essential for crystallization, preparation of heavy-atom derivatives and solution of the non-crystallographic symmetry.
PDB reference: bacteriophage λ lysozyme, 1am7

Re-examination of the known data on crystalline forms of polyglycine reveals that the crystal modification `polyglycine I' has two different three-dimensional structures depending on the molecular weight. Structures for both polyglycine I crystals are described.
The three mutants, (D99A and H48Q) and the calcium-loop mutant (D49E) show that the active site is elongated encompassing the above three regions.
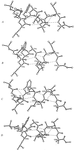
The structure of four conformers of a helical nonapeptide is solved by use of Se substitution, Cu Kα radiation and the linear equations for anomalous dispersion.

A method is described for the determination of monomer envelopes and non-crystallographic symmetry matrices from a solvent-flattened map. A process of envelope skeletonization determines approximate monomer envelopes which are used in conjunction with real- or reciprocal-space techniques to automatically average a map from the solvent-flattening stage.
Torsion-angle molecular dynamics makes real-space methods the most efficient for all but the final cycles of refinement when excellent (anomalous diffraction) phases are available. Overfitting is reduced and convergence improved by implicit use of phases and use of a local refinement that does not allow remote errors to be mutually compensating.
When heavy-atom derivatives contain common sites, improved calculated centroid phases and corresponding figures of merit are obtained by application of the PBR procedure to MIR or MAD data sets: the refinement and phasing calculations are split into two in order to obtain independent events.
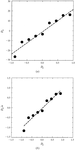
Standard errors in atomic displacement parameters resulting from protein crystal structure determinations are estimated by comparing protein structure pairs of identical sequence.
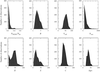
The number of water molecules which are expected to be experimentally located by protein crystallography is determined by analysis of known protein crystal structures.
An efficient new algorithm for six-dimensional molecular-replacement searches is described. The procedure offers several advantages over existing molecular-replacement methods.

The effects of parameter and method variation on the success of the direct-methods procedure Shake-and-Bake are described for six small protein structures.
Open  access
access
access
The presence of distinct regions of high and low density variation in electron-density maps is found to be a good indicator of the correctness of a heavy-atom solution in the MIR and MAD methods.

As the accuracy of protein crystal structure determination improves, protein backbone conformations are observed to be increasingly drawn towards only a few conformational attractors, revealed in novel differential Ramachandran maps.
crystallization papers

Selenomethionine glycinamide ribonucleotide synthetase has been crystallized. The crystals belong to space group P212121 with a = 56.2, b = 62.4 and c = 129.8 Å, and diffract to 1.6 Å resolution at the Cornell High Energy Synchrotron Source.

Two crystal forms of the alpha isoform of rat phosphatidylinositol-transfer protein (PITP-α) with bound phosphatidylcholine have been grown that are suitable for structure determination using X-ray diffraction techniques. Both crystal forms are monoclinic with P2 space group and diffract X-rays to greater than 2.1 Å.
Crystallization of the recombinant monomeric and dimeric B-domain repeats of S. aureus collagen-binding surface protein.
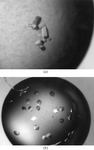
Two different crystal morphologies have been obtained for T. cruzi dUTPase. Complete X-ray diffraction data have been collected for one of the forms.
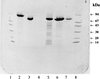
Limited proteolysis was used to produce a dimeric (2 × 70 kDa) crystallizable protein fragment of Escherichia coli pyruvate formate-lyase. The crystals belong to space group P61 or P65 and diffracted to better than 3.2 Å resolution.
A complex of the antibiotic vancomycin with a dipeptide cell-wall analogue crystallizes in an unusually long unit cell, suggestive of molecular stacking.

Two crystal forms of human thioredoxin peroxidase-B have been grown from non-recombinant protein purified from red blood cells. These crystals diffract X-rays to beyond 2.0 Å resolution.
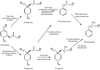
Cyclohexadienyl dehydratase from P. aeruginosa has been crystallized in a new crystal form, which diffracts beyond 1.6 Å at the Cornell High Energy Synchrotron Source (CHESS) A1 beamline.

The molybdate sensor and transcriptional regulator ModE has been crystallized in two forms. The crystals are well ordered and suitable for structure determination.

1,5-α-Arabinanase A from P. fluorescens subspecies cellulosa crystallizes in the hexagonal space group P6122 with unit-cell parameters a = b = 91.6, c = 179.4 Å. Cryocooling conditions have been established, though a shrinkage of the unit cell is observed, with a = b = 88.8 and c = 176.9 Å.
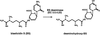
Blasticidin S deaminase from A. terreus was crystallized and the rhombic shaped crystal was subjected to X-ray structure analysis. Preliminary diffraction data were collected to a resolution of 2.0 Å with good statistics.
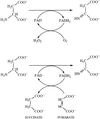
The flavoenzyme L-aspartate oxidase is homologous to the flavoprotein subunit of succinate dehydrogenase and fumarate reductase. Crystals of L-aspartate oxidase from E. coli have been obtained using PEG 4000 as preciptant.

The unusual dodecameric ferritin from Listeria innocua has been crystallized using PEG 1000 as precipitant. The crystals are orthorhombic, space group P212121 and diffract to 2.35 Å.

Recombinant constructs encoding the fibrinogen-binding domains of ClfA and ClfB from Staphylococcus aureus have been crystallized.

Diffraction data have been collected to 3.6 Å resolution from flash-frozen crystals of the T = 4 hepatitis B capsid. Self-rotation functions clearly indicate the orientation of the capsid in the unit cell.
Importin α is the nuclear import receptor that recognizes `classical' nuclear localization sequences. Recombinant mouse importin α has been crystallized and two crystal forms characterized.

The acidic lectin from the winged bean has been crystallized in two forms. Preliminary crystallographic investigations, including molecular replacement, have been carried out.

Crystallization and preliminary X-ray study of chloroplast glyceraldehyde-3-phosphate dehydrogenase
Glyceraldehyde-3-phosphate dehydrogenase from spinach chloroplasts has been crystallized by vapour diffusion. Data collection up to 3 Å has been carried out.
short communications

Pig kidney DOPA decarboxylase, expressed in Escherichia coli, gave rise to crystals of space group P622 with three to five copies of the homodimer in the asymmetric unit. Preliminary X-ray studies to 2.5 Å resolution of this crystal form are reported.
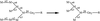
X-ray diffraction quality crystals have been grown and data collected for a key enzyme involved in N-linked oligosaccharide maturation.
Crystals of the active conformation of PAI-1 were grown under different conditions and at least one type was suitable for X-ray diffraction analysis.

Using the implementation of the gel-acupuncture technique, crystal growth rates, as a function of the supersaturation along capillary tubes, were measured.
book reviews
Free