

 journal menu
journal menu





issue contents
December 2000 issue
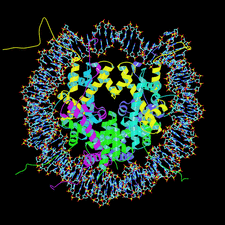
Cover illustration: Nucleosome core particle with the protein represented by ribbon models of the secondary-structure elements and DNA indicating base pairing between complementary strands (p. 1513).
research papers






The structure of the nucleosome core particle at 2.5 Å resolution is presented. Asymmetries found in the structure are examined and discussed along with details of ion binding, hydration and lattice-packing interactions.
PDB reference: nucleosome core particle, 1eqz
A systematic molecular-replacement case study revealed an approximate correlation between the accuracy of an NMR search model and the clarity and quality of the solution. Ensemble models were found to have a larger tolerance in inaccuracy than single averaged models and are more likely to lead to success.
The structures of three R6 hexameric insulin structures are reported, two of which are monoclinic and complexed with either m-cresol or resorcinol, and one rhombohedral complexed with m-cresol.
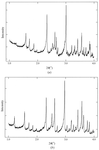
High-resolution synchrotron X-ray powder diffraction data have been used to solve the crystal structure of a new variant T3R3 human insulin–zinc complex produced by mechanical grinding of a polycrystalline sample.
The phases have been determined directly of the 59 kDa protein OppA co-crystallized with uranyl ions.
The crystal structure of the family IIIa cellulose-binding domain (CBD) from the cellulosomal scaffoldin subunit of C. cellulolyticum was determined. The structure proved very similar to the previously elucidated scaffoldin CBD from C. thermocellum, which enabled comparative analysis of possible functional consequences in the shallow groove and cellulose-binding faces among various CBD families.
PDB reference: CBD, 1g43
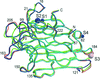
The crystal structures of ZnCa con A at pH 6.15 and CdCd con A at pH 5.0 reveal the role of the crystal environment in the structure of the protein surface. Alternative molecular packings of the con A subunits are established to satisfy the interdependent conditions of pH, quaternary structure and crystal packing. These packings are further stabilized by the formation of supplementary metal-binding sites, S3 and S4.
The crystal structure of cytochrome c6 from the red alga P. yezoensis has been determined at 1.57 Å resolution. The sequence and the structure of the eukaryotic red algal cytochrome c6 are very similar to those of a prokaryotic cyanobacterial cytochrome c6 rather than those of eukaryotic green algae c6 cytochromes.
PDB reference: cytochrome c6, 1gdv

A general approach based on mass spectrometry is described for the rapid identification of the content of macromolecular crystals.
ConfMatch is a global real-space fitting procedure that builds a detailed protein model directly from the initial electron-density map. Experiments have shown that ConfMatch may be able to fully automate the interpretation of density maps of small proteins.
General quadratic functions in real and reciprocal space and their application to likelihood phasing
The properties of a class of quadratic summations are examined under Fourier transformation of the working variables. The relationship with reciprocal-space (likelihood-based) solvent flattening is examined.

The molecular-replacement method has been extended to a simultaneous search for multiple copies of the macromolecule in the unit cell using a special translation function.

Enzymatic deglycosylation and cryoprotection with MPD improved the resolution of agonist-free AMPA receptor ligand-binding domain crystals from 7 to 2.6 Å.
crystallization papers
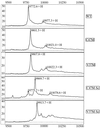
The ribonuclease domain of the ribosome-inactivating colicin E3 in complex with its inhibitor immunity protein has been cystallized. The crystals belong to the space group P3121 or its enantiomorph, with unit-cell parameters a = b = 93.7, c = 76.2 Å. Assuming a single copy of the complex in the asymmetric unit yields a solvent content of 72% and when flash-frozen at 100 K these crystals diffract to a resolution of 2.4 Å.

Several crystal forms of FtsZ, an essential cell-division protein of mycobacteria, are described, including that of a complex with a novel polymerization inhibitor, SRI-7614.

Crystals suitable for structural analysis have been prepared from RPbAI, a legume lectin from R. pseudoacacia.

The quality of a crystal of retinol dehydratase is significantly improved with covalent modification of the protein by ethylmercury.

Crystals of the neuropeptidase neurolysin have been produced and shown to diffract to high resolution. Circular dichroism studies and sequence-based prediction indicate that the enzyme is primarily α-helical.
Tetracenomycin A2 oxygenase catalyzes the final step in the biosynthesis of the polyketide natural product tetracenomycin C. Tetracenomycin A2 oxygenase, which contains an FAD cofactor, catalyzes a novel triple hydroxylation reaction.

The crystallization and preliminary characterization of the MspI restriction endonuclease–DNA complex is reported. Crystals belong to the monoclinic space group and diffract to beyond 2.0 Å.

The structures of both the dehydrogenase and oxidase forms of xanthine oxidoreductase have been determined.

Earthworm fibrinolytic enzyme component A, a protein which functions both as a direct fibrinolytic enzyme and a plasminogen activator, has been crystallized in the orthorhombic space group P212121. The single crystals diffract to 1.95 Å resolution using synchrotron radiation.

The catalytic domain of murine terminal deoxynucleotidyl transferase has been crystallized in its active form. The crystals diffract up to 2.4 Å resolution.
Recombinant L-methionine-α-deamino-γ-mercaptomethane lyase from P. putida has been crystallized. Native data have been collected to 2.68 Å resolution using synchrotron X-rays.

Endopolygalacturonase SE1 from T. penicillatum was crystallized and diffracted to 2.0 Å resolution. The crystals belong to the hexagonal space group P61 or P65, with unit-cell parameters a = b = 135.0, c = 70.7 Å, γ = 120°.

A recombinant anti-17β-oestradiol Fab fragment was overexpressed, purified and crystallized in the presence and absence of oestradiol using microseeding and co-crystallization. The crystals diffracted to 2.0 Å resolution and allowed high-resolution X-ray structure determination.

Adenylylsulfate reductase from A. fulgidus was purified and crystallized under anaerobic conditions. X-ray diffraction measurement revealed that the crystals diffract beyond 2.0 Å resolution.

Crystals of the Y. pestis virulence factor YopM have been obtained by the vapor-diffusion method. The best crystals diffract to 2.4 Å resolution and belong to space group P4222.
Tetrameric protein kinase CK2 holoenzyme from Homo sapiens was successfully crystallized for the first time. The crystals diffract X-rays to at least 3.5 Å resolution.

In this paper, the crystallization, preliminary X-ray analysis and molecular-replacement solution of the carboxy form of haemoglobin I from the fish B. cephalus are described.

The glycosomal enzyme pyruvate phosphate dikinase from T. brucei has been crystallized in space group P21212. A 3.0 Å resolution native data set has been collected at 277 K using monochromatic synchrotron radiation.

GlpE is the first sulfurtransferase composed of a single rhodanese-like domain to be crystallized. Crystals belong to the P31 (or P32) space group, with one molecule per asymmetric unit.

The first E. coli ORFan gene product has been cloned, expressed, purified and crystallized. The MAD method using selenium has been used to solve the selenomethionine-substituted b0220 protein structure.
The R-specific alcohol dehydrogenase from L. brevis was successfully crystallized. The crystals diffract X-rays to at least 1.8 Å resolution.
short communications
Cytochrome c2 from R. palustris has been crystallized in two different crystal forms: monoclinic at pH 4.4 and trigonal at pH 9.0. The preliminary structures show an important change to the iron coordination environment in the trigonal form obtained at basic pH owing to the substitution of the Met ligand by an ammonia molecule.
addenda and errata
Free 






