

 journal menu
journal menu





issue contents
November 2001 issue
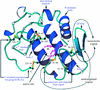
Cover illustration: Manganese ion at the ![[alpha]](/logos/entities/alpha_rmgif.gif) /
/![[beta]](/logos/entities/beta_rmgif.gif) interface of PheRS (p. 1534).
interface of PheRS (p. 1534).
research papers







The mouse L-chain ferritin structure has been refined to 1.6 Å resolution from a cubic F432 crystal.
PDB reference: mouse L-chain ferritin, 1h96

The structure of cytochrome c2 from R. centenum was determined using X-ray diffraction at a resolution limit of 1.7 Å. The implications of the structural features, including a unique extended α-helix and an unusual charge distribution on the surface, are discussed.
PDB reference: cytochrome c2, 1jdl
Polydocanol binds to the hydrophilic regions on the surface of trypsin offering additional stability to the protein.
The crystal structure of U. dioica agglutinin isolectin I was solved by molecular replacement. Two molecules form a pseudosymmetric dimer, mediated by two zinc ions that are bound by residues of the sugar-binding site of the C-terminal domain.
PDB reference: agglutinin isolectin I, 1iqb
The structure of the histidine biosynthetic enzyme HisF from the hyperthermophilic archaeon P. aerophilum has been determined at 2.0 Å resolution. Two tightly bound phosphate ions and two glycerol molecules mark out the probable binding site for the bisphosphoryl substrate of HisF.
PDB reference: HisF, 1h5y
The X-ray crystallographic structure determination of the human liver isozyme of fructose-1,6-bisphosphate aldolase by molecular replacement using a tetramer of the human muscle isozyme as a search model is described.
PDB reference: human liver aldolase, 1qo5
The crystal structure of phenylalanyl-tRNA synthetase from Thermus thermophilus, a class II aminoacyl-tRNA synthetase, complexed with phenylalanyl-adenylate has been determined at 2.6 Å resolution.
PDB reference: PheRS–Phe-AMP complex, 1jjc
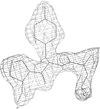
The crystal structure of the murine Tcl1 dimer has been determined at 2.5 Å resolution with an R factor of 0.225. Structural comparisons of the Tcl1 oncoprotein family reveal a high degree of structural conservation, internal symmetry and a common planar surface likely to be involved in protein–protein interactions.
PDB reference: murine Tcl1, 1jnp
The crystal structure of vipoxin, a neurotoxic postsynaptic heterodimeric complex from the venom of Vipera ammodytes meridionalis, has been determined by the molecular-replacement method and refined to 1.4 Å resolution.
PDB reference: vipoxin, 1jlt
The crystal structure of uropepsin isolated from human urine is reported.
PDB reference: uropepsin, 1flh

The structure of an unknown metalloproteinase has been solved with the new direct-methods program ACORN using 1.0 Å resolution data. The whole process, starting from the processed data and ending with a refined model, required less than 6 h of computational time.
PDB reference: deuterolysin, 1eb6
The X-ray crystal structure of two N-terminal integrin-binding IgSF domains of human vascular cell adhesion molecule (VCAM-1) shows an unusual and highly hydrated packing arrangement in which over 80% of the crystal is occupied by solvent. The conformation is also different, with a tilt angle between the two IgSF domains of 19.4°.
PDB reference: VCAM integrin-binding fragment, 1ij9
Eight molecular-dynamics simulations, based on the crystal structures of PNA complexes with two sugars at 293 and 313 K, confirm the qualitative explanation for improved binding of one sugar over the other and provide a rationale for the temperature dependence of thermodynamic parameters and for the observed sugar-binding affinity of one of two available mutants. They also demonstrate the usefulness of carrying out several simulations with different starting conditions. The results of the work may have implications for rational drug-design approaches.
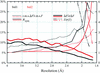
Independent SAD, MAD and MIRAS 1.7 Å phase sets for B. stearothermophilus tryptophanyl-tRNA synthetase show that SAD phasing compares very favourably with the other methods. Maximum-likelihood refinements of the structure against the three sets of experimental amplitudes and phases agree closely, providing significant structural revisions throughout the former 2.9 Å model and documenting subtle localized structural differences between native and selenomethionylated protein.
The destabilizing effect of fluorouracil is compared with the effect of chloro- and bromouracil when they are introduced into a short RNA sequence.
A comparison involving the structures of tetragonal lysozyme grown in the presence of sucrose, sorbitol and trehalose, and those of the orthorhombic and the monoclinic crystals grown under conditions in which tetragonal lysozyme is grown shows that the effect of the additives on the structure and, more importantly, the hydration shell of the protein is less than the normal effect of changes in molecular packing.

The correlation between supersaturation and hen egg-white lysozyme crystal quality was systematically studied employing atomic force microscopy and X-ray crystallography with synchrotron radiation.
crystallization papers

Human dehydroepiandrosterone sulfotransferase is important for the transport of sex-hormone precursors to the peripheral tissues. It has been crystallized in the presence of substrate, of substrate and cofactor, and as an uncomplexed form. The crystals diffract to 2.15, 2.50 and 2.35 Å, respectively.
The DNA-binding domain of the Oct1 transcription factor has been crystallized in the presence of two DNA-response elements. Protein expression, purification, crystallization and crystal mounting were optimized in order to obtain crystals suitable for X-ray analysis.

The xenograft antigen in complex with GS-1-B4 lectin: crystallization and preliminary X-ray analysis
Isolectin B4 of G. simplicifolia lectin-1 has been crystallized in the presence of the xenograft antigen.

Recombinant bacterial cytosine deaminase from Escherichia coli has been subcloned, overexpressed, purified and crystallized for structural analysis. The crystals belong to space group R32, with unit-cell parameters a = b = 109.1, c = 240 Å and diffract to at least 1.5 Å resolution at a synchrotron X-ray source.

The recombinant Peking duck Cu,Zn superoxide dismutase was overexpressed in Escherichia coli, purified to homogeneity and crystallized. In trigonal crystals of the enzyme, unusual supermolecular double-helix packing with 922 non-crystallographic symmetry has been observed in the molecular replacement analysis.

The thermostable β-glycosidase from T. nonproteolyticus HG102 has been cloned and expressed in E. coli. Preliminary crystallographic investigations have been carried out.
Ocr, the product of gene 0.3 of bacteriophage T7, prevents the action of restriction endonucleases of the host bacteria. Ocr has been crystallized in a number of different crystal forms and X-ray data for the seleno-L-methionine-substituted form has been collected to a resolution of 1.8 Å.

c-Myb and the C/EBP family are transcriptional regulatory factors that act in concert to regulate the expression of myeloid-specific genes and v-Myb encoded by avian myeloblastosis virus is an oncogenic mutated form of c-Myb. Crystals of ternary complexes, in which various combinations of the DNA binding domains of c-Myb or v-Myb and C/EBPα or C/EBPβ are bound to a DNA fragment from tom-1A promoter, were obtained by the vapour-diffusion method.

The archaeosine tRNA-guanine transglycosylase from the hyperthermophilic archaeon P. horikoshii was crystallized and data were collected to 2.2 Å. Selenomethionine-labelled protein crystals were prepared, with the aim of solving the structure using a MAD phasing method.

Rapana hemocyanin functional unit RtH2-e was isolated and crystallized. X-ray data to 3.3 Å resolution have been collected.
The psychrophilic enzyme phosphoglycerate kinase from the antarctic bacterium Pseudomonas sp TACII18 has been crystallized alone and in the presence of its product and inhibitor: 3-PGA and AMP-PNP. X-ray diffraction data have been collected on both types of crystals and the cocrystallized data set has been phased by molecular replacement.
The heterodimeric disintegrin with a molecular weight of 14 kDa from E. carinatus venom is a potent antagonist of α4 integrins. Crystals of disintegrin belonging to space group I4122 with unit-cell dimensions a = b = 91.7, c = 55.7, Å have been produced.

The asymmetric unit of the T. cruzi pteridine reductase crystal contains a tetramer related by non-crystallographic 222-symmetry.

Aspartate racemase from P. horikoshii has been expressed, purified and crystallized in three crystal forms. The crystals with space group P21 diffracted to 1.7 Å resolution using synchrotron radiation.
The protegrin-3 prosequence which belongs to the cathelicidin family was overexpressed in E. coli, purified to homogeneity and crystallized. Crystals were shown to diffract beyond 1.9 Å and belong to either space group P6122 or P6522 with unit-cell parameters a = b = 51.42, c = 134.25 Å and have one molecule in the asymmetric unit.

The flavoenzyme acyl-CoA oxidase from rat liver has been crystallized by the hanging-drop vapour-diffusion technique using PEG 20 000 as a precipitating agent. The crystals belong to space group P212121 and diffract beyond 2.5 Å resolution.

Monomeric isocitrate dehydrogenase was crystallized and preliminary X-ray diffraction studies were performed by the MAD method using Mn atoms.

Trp138Phe/Val185Thr xylose isomerases from T. neapolitana and T. thermosulfurigenes have been crystallized in a form suitable for X-ray diffaction studies. The crystals belong to the orthorhombic space groups C2221 (TNXI mutant) and P212121 (TTXI mutant).

The C-terminal domain of the HrcQB protein from P. syringae was overexpressed, purified and crystallized. Native X-ray diffraction data were collected to 3.0 Å using a laboratory source.

TB-RBP temporally and spatially controls mRNA translation; the protein has been crystallized in space group P21212.

The replication-fork regression helicase recG from T. maritima has been crystallized in complex with a three-way DNA junction.

Three isozymes of frog ribonucleases from R. catesbeiana oocytes have been crystallized under different conditions in various space groups.

Crystals of an antigen-binding fragment of the mouse anti-human Fas monoclonal antibody HFE7A have been obtained. Fast optimization to produce single crystals suitable for X-ray analysis was achieved by the streak-seeding technique.

The 3B62 antibody Fv expressed in the yeast P. pastoris was secreted as a heterodimer consisting of the VL and VH domains. It retains nitrophenol-binding affinity comparable with that of the 3B62 Fab and yields crystals of good diffraction quality.
Uracil-DNA glycosylase (UDG) is a DNA-repair enzyme involved in the removal of uracil from DNA. Atlantic cod UDG has been crystallized by the vapour-diffusion method using sodium citrate as the precipitant at pH 7.5.
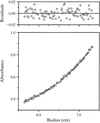
Uroporphyrinogen-III decarboxylase from the tobacco plant is a plastidial enzyme involved in the biosynthesis of chlorophyll and haem. Sedimentation equilibrium and dynamic light scattering revealed a dimeric form in solution. Good quality diffraction crystals were obtained using synchrotron radiation.

Crystals of the catalytic domain of a human thioredoxin-like protein have been obtained by the vapour-diffusion method. The results of the preliminary X-ray diffraction analysis of these crystals are discussed.

A molybdate/tungstate binding protein has been crystallized in the presence and absence of oxyanion. High-resolution diffraction data have been recorded.

An arsenate reductase from B. subtilis has been expressed, purified and crystallized. Data in space group P212121 have been collected to high resolution from crystals of both wild-type and SeMet proteins.

The negative transcriptional regulator NmrA which is responsible for nitrogen-metabolite repression in certain fungi has been expressed and purified. Three crystal forms have been obtained, two of which appear suitable for high-resolution structure determination.

Glutathione-dependent dehydroascorbate reductase from spinach chloroplasts has been crystallized. The crystals diffract to 2.2 Å and belong to space group C2.
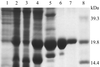
recombinant tabtoxin-resistance protein complexed with acetyl-coA as well as its selenomethionine derivative were crystallized. The crystals from the derivative were found to belong to two crystal forms and diffracted to high resolution.

Crystals of the quinohaemoprotein alcohol dehydrogenase from C. testosteroni were obtained and a 2.44 Å resolution data set has been collected. An approximate orientation of the haem groups has been derived by inspection of the crystals using plane-polarized light.
short communications
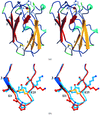
A surface disulfide has been engineered in popular plastocyanin, at residues 21–25. The site is at about 20 Å from plastocyanin type I copper, whose coordination sphere is not affected by the engineered disulfide. The mutant plastocyanin has been successfully employed for chemisorption on gold substrates.
PDB reference: plastocyanin, 1jxg
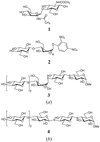
The 1.1 Å structure of a retaining endocellulase, Cel5A, in complex with methyl 4,4II,4III,4IV-tetrathio-α-cellopentoside reveals the binding of five pyranosides, all in the 4C1 chair conformation, occupying the −3, −2, +1 and +2 subsites.
PDB reference: Cel5A–SDP5 complex, 1h5v

The binding site of doxorubicin is identified from crystal structure analysis.
PDB reference: BoNT/B–doxorubicin complex, 1i1e

Structure of YlxR homologue from S. pneumonia was determined using MAD phasing and synchrotron radiation. The protein shows a new fold.
PDB reference: YlxR, 1g2r

ADP is a fully automated data-processing program for protein crystallography. It performs analysis and makes choices the way crystallographers usually do on a beamline when using standard integration software.
addenda and errata
Free 






