

 journal menu
journal menu





issue contents
June 2002 issue
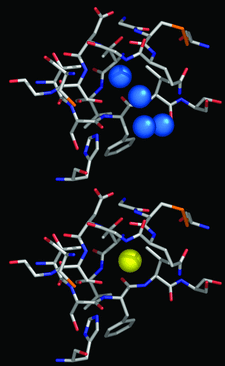
Cover illustration: Region of the elastase molecule showing the common binding site for iodine (top) and xenon (bottom), p. 976.
lead articles




Automation of crystal optimization techniques for adaptation to high-throughput techniques is described.
research papers
The probabilistic estimate of the amplitudes for normal scattering from the anomalous scattering substructure is attempted in the two-wavelength case.



The structure of uracil phosphoribosyltransferase from B. caldolyticus with bound UMP is described and a catalytic mechanism is proposed.
PDB reference: uracil phosphoribosyltransferase, 1i5e, r1i5esf
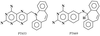
Structural data are reported for N-(2,4-diaminopteridin-6-yl)methyldibenz[b,f]azepine (PT653), an example of structure-based inhibitor design with 21-fold selectivity for P. carinii dihydrofolate reductase (pcDHFR) relative to rat liver dihydrofolate reductase (rlDHFR).
PDB reference: NADPH–PT653–pcDHFR, 1klk, r1klksf
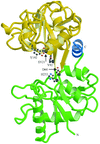
The structure of the R121D mutant of the N-lobe half-molecule of human lactoferrin has been determined and refined at 3.0 Å resolution using data from a merohedrally twinned crystal form. The mutant, in its iron-free form, has a domain-opened structure that matches that of the N-lobe of full-length human lactoferrin.
PDB reference: lactoferrin, 1l5t, r1l5tsf
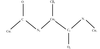
A method is presented for high-resolution protein crystallographic map interpretation.
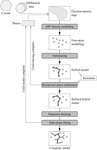
Algorithms for automatic model-building in ARP/wARP.
In-house protein derivatization and structure solution using triiodide solutions is illustrated using five test cases. Also discussed is the method of combinatorial counter-ion replacement and its usefulness on conventional X-ray sources.
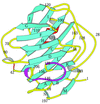
The structure of calcium-depleted human C-reactive protein has been determined at 3.15 Å from perfectly twinned crystals. The structure reveals two independent pentamers which form a face-to-face decamer across a dyad near-parallel to the twinning twofold axis.
PDB reference: C-reactive protein, 1lj7
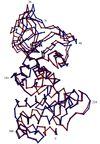
The structure of thermolysin in the absence of substrate has been determined in a new crystal form. The enzyme takes an open conformation consistent with hinge-bending during the catalytic cycle.
PDB reference: tetragonal thermolysin, 1l3f, r1l3fsf
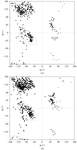
The crystal structures of pectate lyase A from E. chrysanthemi EC16 are reported in two space groups, C2 and R3. Comparison to other pectate lyase isoforms from E. chrysanthemi is discussed.
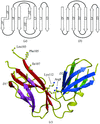
The 2.2 Å crystal structure of the periplasmic chaperone SfaE is described and compared with a number of previously determined structures of homologous chaperones.
PDB reference: SfaE, 1l4i, r1l4isf
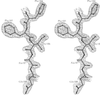
The structure of ribosomal protein L1 from M. thermolithotrophicus compared with L1 protein structures from two other sources reveals two regions which retain their structure in all three proteins. These structural invariants are involved in intermolecular interactions in the crystals and could be treated as potential RNA-binding sites.
PDB reference: ribosomal protein MthL1, 1dwu, r1dwusf
crystallization papers

To investigate how NBD2 participates in ClpB function to disaggregate denatured proteins, ClpB NBD2 has been cloned and crystallized.

Crystallization conditions and X-ray diffraction data for a new form of concanavalin A are described.

BaP1, a venom haemorrhagic class P1 metalloproteinase, has been crystallized and diffraction data have been collected to 2.7 Å resolution.

Bothrombin, a serine protease which activates blood coagulation factor VIII, aggregates platelets and cleaves fibrinogen, has been crystallized and diffraction data have been collected to 2.8 Å resolution.

A new refolding procedure has facilitated growth of diffracting crystals of elongation factor P in the absence of contaminating oligonuleotides.

Expression, purification and crystallization of a collagen-binding fragment of Yersinia adhesin YadA
A recombinant collagen-binding fragment of adhesin YadA of Y. enterocolitica serotype O:3 has been produced in E. coli, purified and crystallized.

N-Acetyl-L-glutamate kinase from P. aeruginosa catalyses the controlling step of arginine biosynthesis and is feedback inhibited by arginine. The gene for this enzyme was cloned and expressed at high levels in E. coli. Crystals of the purified recombinant enzyme, belonging to space group P1, diffracted to a resolution of 2.75 Å.

The 17β-hydroxysteroid dehydrogenase type 5 responsible for androgen biosynthesis in peripheral tissues has now yielded high-resolution crystals.

The chromo shadow domain (CSD) of the murin HP1-like protein M31 has been crystallized and X-ray diffraction data have been collected to 2.9 Å. The native crystals belong to space group C2221 with unit-cell parameters a = 60.0, b = 95.6, c = 91.7 Å and α = β = γ = 90°

Crystals of acylamino acid releasing enzyme from hyperthermophilic archaeon A. pernix strain K1 have been obtained. The crystal belongs to the space group P1 with unit cell dimensions of a =107.5, b =109.9 and c =119.4 Å, α =108.1, β= 109.8 and γ = 91.9° at 2.9 Å resolution.
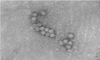
Crystals of the JX/78 strain of SVDV were obtained from virus in two wells of crystallization conditions and present preliminary X-ray data to 3.6 Å resolution.
Leucine dehydrogenase from T. intermedius has been crystallized as a binary complex with 2-ketoisocaproate and as a non-productive ternary complex with NAD+ and 2-ketoisocaproate in forms suitable for structure determination.

The PDZ6 domain of GRIP1 from R. norvegicus has been crystallized in the apo form and complexed with the octapeptide of the liprin-α1 C-terminus. The native and complex crystals belong to space group P6122 (or P6522) and R32 and diffracted to 1.5 and 1.8 Å, respectively.

The ATPase domain of the membrane-anchored protease FtsH of E. coli has been crystallized and native data to 1.5 Å spacing have been collected using synchrotron radiation.
A truncated form of brefeldin A-ADP ribosylated substrate (residues 1–350), a protein involved in Golgi membrane fission, has been expressed, purified and crystallized. SeMet-substituted crystals were also prepared, aiming at the three-dimensional structure solution via MAD methods.
Enoyl-ACP reductase from H. pylori has been crystallized in the presence of its cofactor NADH and the inhibitor triclosan (or its analogue diclosan). Diffraction data have been collected to 2.5 Å (or 2.3 Å) resolution with synchrotron X-rays.
4-Chlorocatechol 1,2-dioxygenase from the Gram-positive bacterium R. opacus (erythropolis) 1CP, an enzyme involved in the aerobic biodegradation of chloroaromatic compounds, has been crystallized. This is the first intradiol dioxygenase which specifically catalyzes the cleavage of chlorocatechols in Gram-positive bacteria to give diffraction-quality crystals.
short communications

The crystal structure of a mouse phosphatidylethanolamine-binding protein homologue (mPEBP-2) has been determined to 1.8 Å. Regions of distinctive sequence associated with the PEBP-2 subset identified from alignments of the PEBP family are discussed with reference to the structure.
PDB reference: mPEBP2, 1kn3, r1kn3

High-temperature incubation and iodination of a specific tyrosine residue are required for crystallization and SAS structure determination of an antifreeze protein. These methods can be employed to modify other target proteins in order to aid in their crystallization and structure determination.
book reviews
Free 






