

 journal menu
journal menu





issue contents
December 2003 issue
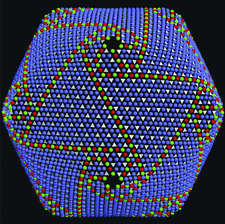
Cover illustration: Color-coded distribution of three classes of intercapsomer contacts in the 1900 Å diamoeter dsDNA Paramecium bursaria Chlorella virus. The pseudo hexagonal trimeric capsomers are centered around the white triangles (p. 2053).
editorial




Free 

10th anniversary papers

An analysis is presented of pseudo-hexagonal capsomer arrays that make up the structure of very large icosahedral dsDNA viruses.
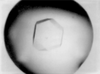
Prohead II is ∼50 nm in diameter and is the first assembly intermediate of a dsDNA bacteriophage capsid to be crystallized. The crystals diffract to 5 Å resolution and the crystal packing of this icosahedral early intermediate of HK97 in the P213, 707 Å unit cell is reported.


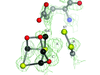
Crystals prepared from an old sample of the hybrid cluster protein (HCP) from D. desulfuricans ATCC 27774 contain molecules in both the as-isolated oxidized and reduced states.
PDB reference: hybrid cluster protein, 1upx, r1upxsf

Biological crystallography, spectroscopy, solution X-ray scattering and microscopy have been applied to study the molecular basis of the colouration in lobster shell and this article presents a review of this progress, concentrating on recent results but spanning more than 50 years of work.
research papers

First low-resolution direct phasing of a unknown protein with GENMEM, a program based on the topological analysis of the electronic density. A first simple step of a protein structure determination by X-ray crystallography in a tricky case is described.

The space-group ambiguity for the first plant polygalacturonase to be crystallized, tomato PG2, is discussed and resolved.

With the recent development of accurate implicit solvent models, it is time to test whether electrostatic forces, including the contributions of crystal solvent, should be re-introduced into protein X-ray structure refinements. Using the Generalized Born continuum solvent model, promising results are obtained for three proteins solved at moderate resolutions.
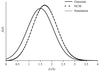
The analytical expression for the coordinate error-dependent distribution of an interatomic distance and a simple relation between the positional error and the map correlation are presented, which can be used in assisting the decision-making process during automated model-building procedures.
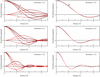
In this account, the use of Fisher's information matrix and the related scoring method of minimization in maximum-likelihood macromolecular crystallographic refinement is discussed. The scoring method has been implemented in the program REFMAC5 from CCP4. The algorithm uses the sparse approximation of the matrix and is computationally very efficient.
The structure of viscotoxin A3 was solved by sulfur-SAD phasing using Cu Kα radiation and an innovation in the program SHELXD for the direct location of disulfide units.
PDB reference: viscotoxin A3, 1okh, r1okhsf
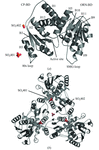
The three-dimensional crystal structure of the triple-point mutant of the catalytic subunit of human protein kinase CK2α has been determined at 2.4 Å resolution. Microcrystals were obtained by a protein-crystallization method based on thin-film nanotechnology and diffraction data were collected by the microfocus beamline at the ESRF synchrotron.
PDB reference: human protein kinase CK2, 1na7, r1na7sf
The high-resolution (1.87 Å) structure of dodecameric ornithine carbamoyltransferase from the hyperthermophilic archaeon P. furiosus allows a finer description of the active site and of the interactions between the different interfaces from which the hyperthermostability of the protein emerges.
PDB reference: ornithine carbamoyltransferase, 1pvv, r1pvvsf
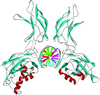
Topological analyses of medium-resolution electron-density maps of macromolecules allow critical point graphs to be obtained which are reduced representations of the structures, including physico-chemical properties. The method has been applied to two protein–DNA complexes, NF-κB and HIF-1.
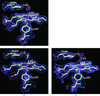
The structure of P. mirabilis catalase in its ground state, oxidized state and in complex with an inhibitor, formic acid, has been solved at 2.3 Å resolution.

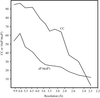
The reflection profiles from single-domain and multi-mosaic domain insulin crystals were analyzed using highly monochromatic parallel synchrotron radiation and fine φ-slicing to help understand the effects of temperature on diffraction quality. Corresponding effects on the insulin structure were re-examined.
Crystal structures are presented from two cyanobacterial forms of the protein PII. Their overall structures are remarkably similar to that of other known PII proteins despite the fact that they mediate different biochemical processes, cell responses, and stimulate a different set of receptors.

The crystal structure of superoxide dismutase from P. aerophilum was determined by molecular replacement at 1.8 Å resolution. Overcoming the difficulties in detecting twinning in the presence of non-crystallographic symmetry is discussed as well as the difficulties in molecular replacement with multiple molecules and pseudo-crystallographic symmetry.
PDB reference: P. aerophilum superoxide dismutase, 1p7g, r1p7gsf
A public web service for effective map improvement, bias removal and structure validation has been implemented on a Linux multi-CPU cluster.

The crystal structure of an insect δ-class glutathione S-transferase agGSTd1-6 from a DDT-resistant strain of the malaria vector A. gambiae has been determined at a resolution of 2.0 Å.
PDB reference: agGSTd1-6, 1pn9, r1pn9sf
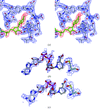
The crystal structure of rabbit-muscle GAPDH at 2.4 Å, with one NAD+ molecule bound to each dimer of the tetrameric enzyme, is the first detailed structure of a mammalian GAPDH and could aid in structure-based drug design.

Water reconstruction during a low-temperature phase transition in flash-cooled concanavalin A is described. The phase transition and its implications for controlled annealing of protein crystals are discussed.

The structure of the α-amino acid ester hydrolase from A. turbidans has been determined. A description is given of the structure-solution process, which was severely hampered by the simultaneous occurrence of twinning and pseudo-translational symmetry.
structural genomics papers

A protein of unknown function, PAE2307, from the hyperthermophilic archaeon P. aerophilum has been crystallized and gel screening has been used to identify a suitable derivative for SAD phasing, given only two usable crystals.
crystallization papers
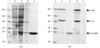
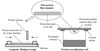
This study describes the crystallization and X-ray analysis of the extracellular region of the human IgA receptor FcαRI. X-ray diffraction data were collected to a limiting resolution of 2.8 Å using synchrotron radiation.

Expression, refolding, crystallization and MAD data collection of the extracellular fragment of FcαRI/CD89 is reported.

The DNA-binding protein from stationary phase cells (Dps) of M. smegmatis has been crystallized in three different crystal forms.

The first crystallographic study of an α-amylase from an organism that is both thermophilic and halophilic, at 1.97 Å resolution is reported.

The extracellular region of NKp46 (22.7 kDa), a natural cytotoxicity receptor composed of two Ig-like domains held to be involved in cellular ligand recognition and binding, has been crystallized in a hexagonal crystal form suitable for a detailed structural investigation.

The Holliday junction resolvase enzyme RusA from E. coli has been purified and crystallized. A selenomethionine-incorporated form of the enzyme that diffracts to 2.1 Å resolution has been used to collect a full three-wavelength MAD data set.

Crystals suitable for structural analysis of octaprenyl pyrophosphate synthase, a long-chain trans-type prenyltransferase, have been obtained from thermophilic T. maritima and mesophilic E. coli. The structure of the T. maritima enzyme has been solved at 2.28 Å resolution.

The C-terminal domain of human FKBP52 has been crystallized. The crystals diffracted to 2.7 Å.

S-Formylglutathione hydrolase has been crystallized and several crystallographic data sets have been collected to a maximum resolution of 1.6 Å. The space group changes from monoclinic C2 to trigonal P3121 or P3221 when the crystal is pressurized in a xenon pressure chamber.

The small component of 4-hydroxyphenylacetate 3-monooxygenase (HpaC) from T. thermophilus HB8 has been crystallized in both apoprotein and flavin-complex forms, and these crystals diffracted to 1.85 and 1.3 Å resolution, respectively.

The cell-wall invertase inhibitor Nt-CIF from N. tabacum has been expressed in E. coli. Crystallization experiments of the purified protein resulted in four different crystal forms, all diffracting to resolutions higher than 2.5 Å.
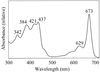
A water-soluble chlorophyll protein (WSCP) with a chlorophyll a:b ratio of 6:1 from B. oleracea L. var. acephala (kale) was purified and crystallized. A native data set was collected to 2.8 Å resolution at 293 K using Cu Kα radiation from a rotating-anode generator.
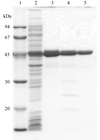
A recombinant form of A. globiformis inulin fructotransferase was overexpressed in E. coli, purified to homogeneity and crystallized by the hanging-drop vapour-diffusion technique using lithium sulfate as a precipitant. The crystal diffracted to better than 1.5 Å at 100 K.

α-Galactosidase I from M. vinacea was crystallized in two tetragonal forms by the hanging-drop vapour-diffusion method.
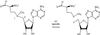
The purification and crystallization of an enzyme from S. cattleya which is responsible for the synthesis of the C—F bond during fluoroacetate and 4-fluorothreonine biosynthesis is reported.

Crystals of the 3-hydroxyisobutyrate dehydrogenase from T. thermophilus HB8 have been obtained and diffract to 1.8 Å resolution. Crystals belong to space group P212121, with a tetramer in the asymmetric unit.

L-Serine dehydratase from human liver was overexpressed in E. coli, purified and crystallized. A complete native data set to 2.5 Å resolution has been collected and structure determination is in progress.

Deliberate protein oxidation has led to bigger crystals and additional crystal forms of the IB1-SH3 domain. The rectangular prism form diffracts to 3.0 Å resolution.

Crystallization and preliminary X-ray diffraction data of Mycobacterium tuberculosis FbpC1 (Rv3803c)
The crystallization of M. tuberculosis FbpC1, a secreted protein homologous to the essential antigen 85 complex but of unknown function, is described. Crystals of FbpC1 belong to space group P41212 at room temperature and undergo a temperature-induced phase transition to P212121 upon freezing.

A large single crystal of DsrD (1.7 mm3) has been successfully crystallized and neutron diffraction data were collected to 2.4 Å resolution.
Acucetin, containing both disintegrin-like and cysteine-rich domains, was purified from the autoproteolytic products of a P-III-type metalloproteinase AaH IV. The N-terminal amino-acid sequence was determined and preliminary crystallographic research was carried out.

CprB, an autoregulator-receptor protein derived from S. coelicolor A3(2), was crystallized. MAD data collection at 2.4 Å resolution was carried out at BL41XU, SPring-8.

The deacetylase MshB from M. tuberculosis (Rv1170), which is involved in the biosynthesis of the key antioxidant mycothiol, has been expressed, purified and crystallized in a form suitable for high-resolution X-ray structural analysis.
Phosphoserine aminotransferase from the obligate alkalophile B. alcalophilus was expressed, purified and crystallized in two different space groups, C2221 and P21212. Crystals diffract to 1.7 and 1.6 Å resolution, respectively.

Recombinant glycogen synthase from P. abyssi has been crystallized. Crystals diffracted to 3.5 Å resolution.

Crystals of P. furiosus ferredoxin containing an [Fe3S4]-cluster have been prepared and X-ray diffraction data have been collected to 1.5 Å.

Archaeal alanine dehydrogenases (AlaDehyds) belong to a family that includes bacterial ornithine cyclodeaminase, marsupial lens proteins, and a human thyroxine carrier, but are unrelated to bacterial AlaDehyds. Crystals of the dimeric AlaDehyd from A. fulgidus undergo a space group change on derivatization, leading to structure determination by MAD methods.

Monoclinic crystals of human peptidylarginine deiminase V have been obtained. Native data have been processed at 2.7 Å resolution using the SPring-8 synchrotron-radiation source.

ClpB from T. thermophilus has been crystallized in the presence of adenosine 5′-(β,γ-imido)triphosphate and adenosine 5′-(γ-thio)triphosphate, respectively. This is the first report of the crystallization of a full-length member of the ClpB/Hsp104 family of molecular chaperones.
short communications

Crystals have been grown of protein P4, the hexameric packaging NTPase, for several dsRNA bacteriophages. Comparison of the packing arrangements of different P4 hexamers and analysis of the disorder provides insights into the, probably biologically relevant, flexibility of this family of proteins.
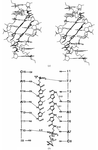
The crystal structure of d(ITITACAC) complexed with distamycin illustrates the features of the 2:1 side-by-side binding mode adopted by alternating DNA octamer duplexes.
PDB reference: 1jux, r1juxsf
NDB reference: d(ITITACAC)–distamycin complex, DD0043

A new method is used to show that the detergent dodecyl maltoside does not migrate into the oil in a microbatch experiment and may be safely used in microbatch crystallization experiments.
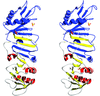
The molecules of E. coli MobB, an enzyme involved in the final step of molybdenum cofactor biosynthesis, form intertwined dimers. These dimers assemble into helices, two of which form a hollow cylinder with a diameter of 250 Å and pitch of 98 Å. These cylinders pack into a hexagonal lattice with P6422 symmetry and 75% solvent content.
PDB reference: MobB, 1p9n, r1p9nsf

The control of detergent-to-protein ratio using Bio-Beads was necessary to obtain crystals that diffract to 2.2 Å resolution. Two crystal forms were obtained, one of which presented a defect that is analyzed herein.

Malonate: a versatile cryoprotectant and stabilizing solution for salt-grown macromolecular crystals
Data supporting the application of sodium malonate as a universal cryoprotectant and stabilizing solution for salt-grown protein crystals are presented.





