

 journal menu
journal menu





issue contents
February 2004 issue
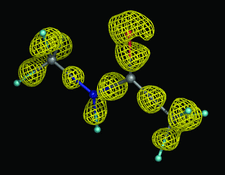
Cover illustration: Refinement at 0.6 Å resolution makes the deformation density visible in conventional macromolecular difference maps (p. 260).
10th anniversary papers






The rigid and the flexible regions of the peanut lectin molecule have been delineated and the invariant water molecules in its hydration shell identified using the structures of the lectin crystals containing ligand-bound and ligand-free subunits, grown under different environmental conditions. The molecule is sturdy and the combining site is essentially preformed.
PDB references: peanut lectin–lactose complex at acidic pH in cacodylate buffer, 1v6i, r1v6isf; in acetate buffer, 1v6j, r1v6jsf; in the presence of oligopeptide 9PG (PVIWSSATG), 1v6k, r1v6ksf; in the presence of oligopeptide 9IA (IWSSAGNVA), 1v6l, r1v6lsf; native peanut lectin in the presence of oligopeptide 9IA, 1v6m, rlv6msf; in the presence of oligopeptide 9PG, 1v6n, r1v6nsf; in the presence of oligopeptide 10PG (PVRIWSSATG), 1v6o, r1v6osf
research papers
A relation between a Gaussian perturbation of the atomic positional parameters and the average squared structure-factor amplitude for the estimation of the quality of an atomic model and corresponding phases is presented.
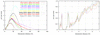
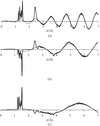
A structural interpretation of 〈|E|2〉(d*) curves is presented and their use for improved parameter estimation in scaling is discussed.
Spallation neutron diffraction data have been measured to better than 2.0 Å resolution on the metalloenzyme D-xylose isomerase at the Protein Crystallography Station beamline at Los Alamos Neutron Science Center. This represents one of the largest proteins and unit cells (∼106 Å3) to be studied to this resolution by neutron diffraction.

The previously unknown structure of a 64-residue fungal protein, denoted the bubble protein, was solved in a highly automated fashion using the anomalous scattering from eight S atoms in data collected on in-house equipment.
PDB reference: bubble protein, 1uoy, r1uoysf
The crystal structure of the V8 protease from S. aureus is reported. From modeling studies, the residues of the enzyme involved in determining its unique specificity for acidic amino-acid residues are predicted.
PDB reference: V8 protease, 1qy6, r1qy6sf
Conventional macromolecular refinement performed at a resolution near 0.6 Å allows regular bond-electron peaks to be observed in difference maps for individual bonds in well ordered molecular domains. These peaks are not visible in the maps if the refinement is performed at a resolution close to 0.9 Å.
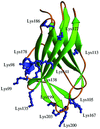
As part of an ongoing effort, the usefulness of Lys→Arg surface mutations in the globular domain of human RhoGDI is studied.
PDB reference: RhoGDI K(199,200)R double mutant, 1qvy, r1qvysf

The crystal structure of the meso-tetrasulphonatophenylporphyrin (H2TPPS)-jacalin complex, determined at 1.8 Å resolution, exhibits binding of the pluripotent porphyrin molecule exploiting the plasticity of the carbohydrate binding site.
The crystal structure of the core region of the soybean β-conglycinin α′ subunit has been determined at 2.3 Å resolution. The structural factors accounting for the difference in thermal stability between the subunits of β-conglycinin are discussed.
PDB reference: soybean β-conglycinin α′-subunit core region, 1uik, r1uiksf

The single Fe atom in the Fe(S-Cys)4 site of C. pasteurianum rubredoxin was exchanged in turn for Cd, Hg, Co, Ni and Ga and the structures of these derivatives were refined.
Structure of circularly permuted DsbAQ100T99: preserved global fold and local structural adjustments
The X-ray structure of a circularly permuted variant of thiol-disulfide oxidoreductase, cpDsbAQ100T99, is reported in which the natural termini are joined by the pentapeptide linker GGGTG, leading to a continuous thioredoxin domain, and new termini that have been introduced in the helical domain by breaking the peptide bond Thr99-Gln100.
PDB reference: DsbAQ100T99, 1un2, r1un2sf
crystallization papers

The S component of the Panton–Valentine leucocidin from S. aureus has been crystallized. A complete native data set to 2.1 Å resolution has been collected.
The construct design by limited proteolysis, subsequent cloning, purification and crystallization of the PH domain of human PDK1 is reported, along with a Patterson map of a gold-derivatized crystal.

The crystallization of rat monoamine oxidase A and general considerations for the crystallization of membrane proteins are presented.
The helicase fragment of Vasa was crystallized in complex with poly(U) RNA and a non-hydrolyzable analogue of ATP. The crystal diffracted to 2.2 Å resolution.

Preliminary crystallographic analysis of the gene-regulatory protein C.AhdI from the bacterial restriction-modification system A. hydrophilia has been undertaken. Native and selenomethionine-derivative data were collected to resolutions of 2.2 and 1.7 Å, respectively.

An extended-spectrum β-lactamase, OXY-1a, from K. oxytoca has been crystallized in two crystal forms which diffracted to different resolutions. The orthorhombic crystal diffracted to at least 1.9 Å and is thus suitable for high-resolution structural studies.

YloQ, a putative GTPase essential for the viability of B. subtilis, has been purified and crystallized. A complete diffraction data set to 2.5 Å spacing has been collected using synchrotron radiation.

Two different crystal forms have been obtained of the fully oxidized form of the dihaem cytochrome c peroxidase from P. denitrificans. One of them diffracts X-rays beyond 2.3 Å resolution and is suitable for structure determination.

The PTB domain of mouse dok1 has been purified and crystallized. A selenomethionmine-substituted protein that diffracts to 2.5 Å resolution has been used to collect a full three-wavelength MAD data set.

Recombinant Ohr was expressed in E. coli as a His6-tagged fusion protein, purified by nickel-affinity chromatography and crystallized using PEG 4000 as precipitant after treatment with t-butyl hydroperoxide. Crystals belong to space group P6522, with unit-cell parameters a = b = 87.66, c = 160.28 Å, and diffract X-rays to a resolution of 1.8 Å.
A novel orange fluorescent protein from the Cnidaria tube anemone Cerianthus sp. was crystallized in space group R3, with hexagonal unit-cell parameters a = b = 216.947, c = 51.839 Å. A set of complete diffraction data was collected to 2.0 Å.

A crystal of mature xylanase B from C. stercorarium has been prepared and the X-ray diffraction data have been collected to 1.80 Å resolution.

Native crystals of recombinant fucosyltransferase NodZ from Bradyrhizobium sp. WM9 (Lupinus) diffract X-rays to 2.85 Å resolution at 100 K. The crystals are hexagonal, space group P6122 or P6522, with unit-cell parameters a = 125.5, c = 95.6 Å, and contain one protein molecule in the asymmetric unit.

Crystals of the human-specific toxin intermedilysin from S. intermedius have been obtained by manipulating the dialysis conditions prior to crystallization.
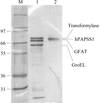
Recombinant purified full-length human PAPS synthetase 1 migrates as a dimer in solution. Monoclinic crystal diffraction intensities have been measured to 1.75 Å resolution using synchrotron radiation.
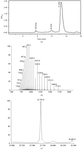
The crystallization and preliminary crystallographic analysis of M. arthritidis-derived mitogen complexed with peptide/MHC class II antigen is described.

The osmotically inducible protein C from T. thermophilus was overexpressed, purified and crystallized. Data were collected at five wavelengths for MAD phasing and the unit-cell contents were analyzed.
An open reading frame from S. pneumoniae 110K/70, a penicillin-sensitive strain, has been cloned, expressed and purified. Crystals obtained from the purified recombinant enzyme have been obtained in two different forms, which may represent different liganded states, diffracting to 2.7 and 1.5 Å, respectively.

Sylvaticin, a protein of the elicitin family, has been crystallized by the vapour-diffusion method and diffracts to 2.1 Å resolution.

The E. coli ytfG gene product, with NAD(P)H:quinone oxidoreductase activity, has been crystallized by the hanging-drop vapour-diffusion method at 296 K.
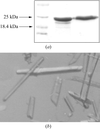
The ribosome recycling factor from M. tuberculosis has been crystallized and the crystals have been characterized.

A mutant of the catalytic domain of mycobacterial adenylyl cyclase Rv1625c has been overexpressed, purified and crystallized. X-ray diffraction data were collected to 3.4 Å resolution.

Using purified, recombinant rat choline acetyltransferase crystals that diffract X-rays to 1.55 Å resolution were generated.

The R2 protein of ribonucleotide reductase from C. trachomatis has been crystallized. Co-crystallization with lead was necessary to obtain crystals.
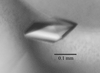
RecR from D. radiodurans has been crystallized (space group C2221; unit-cell parameters a = 106.96, b = 122.25, c = 156.01 Å). Diffraction data have been collected to 2.90 Å using Cu Kα X-rays.

CMY-1 and CMY-10, the plasmid-encoded class C β-lactamases with extended substrate spectrum, were purified and crystallized.
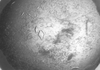
The unique carboxylating enzyme acetone carboxylase from X. autotropicus strain Py2 has been crystallized in the native state and 3.2 Å resolution data have been collected.
short communications
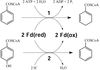
Crystallization of 4-hydroxybenzoyl-CoA reductase and the structure of its electron donor ferredoxin
4-Hydroxybenzoyl-CoA reductase from Thauera aromatica and its electron donor ferredoxin were anaerobically crystallized. The structure of the ferredoxin was solved at 2.9 Å resolution.
PDB reference: ferredoxin TaFd, 1rgv, 1rgvsf
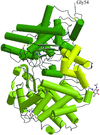
The crystal structures of the soluble extracellular domain of human neutral endopeptidase (residues 52–749) complexed with various potent and competitive inhibitors are described.
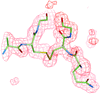
The crystal structure of diaminopimelate epimerase has been refined to 1.75 Å, highlighting differences from the previously determined structure and enabling mechanistic deductions and comparisons with the functionally related amino-acid racemases to be made.
PDB reference: diaminopimelate epimerase, 1gqz, r1gqzsf
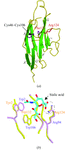
p75/AIRM1, also known as Siglec-7, is a sialoadhesin expressed on natural killer cells that is able to inhibit their cytolytic activity. The X-ray crystal structure of the extracellular IgV-like domain, thought to be involved in target-cell recognition, has been determined at higher resolution (1.45 Å) and in a different space group to the previously reported Siglec-7 structure.
PDB reference: p75/AIRM1 saccharide-binding domain, 1nko, r1nkosf
addenda and errata
Free 






