journal menu
journal menu





issue contents
March 2006 issue
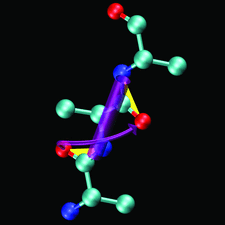
Cover illustration: The figure illustrates the screw motion relating two consecutive peptide planes in a model peptide according to the theorem of Chasles (p. 302).
research papers





Elimination of the strongest correlations between test-set and working-set reflections reduces Rfree bias to acceptable levels when non-crystallographic symmetry is low-order. Bias can not be completely eliminated in the presence of high-order non-crystallographic symmetry, but bias reduction allows optimization of the weights for stereochemically restrained refinement.

The method presented here evaluates crystalline objects in crystallization droplets.


The crystal structure of P. malariae plasmepsin 4 (PmPM4) complexed with the allophenylnorstatine-based inhibitor, KNI-764, is reported. The inhibitor is shown to be bound to PmPM4 in an unexpected orientation with allophenylnorstatine occupying the S1′ pocket.
PDB reference: P. malariae plasmepsin 4, 2anl, r2anlsf
The combined use of non-fluorescent sample holders and short UV-laser pulses to excite the fluorescence of aromatic residues is an efficient tool to visualize protein crystals mounted on synchrotron beamlines. Putative structural damage induced by the short UV-laser pulses was not observed.

The crystal structure of a previously unidentified amylase (AmyC) from the hyperthermophilic organism Thermotoga maritima has been determined at 2.2 Å resolution by means of MAD.
PDB reference: AmyC, 2b5d, r2b5dsf

A new classification system is developed to automatically identify crystal growth in images with a very low false-negative and a moderate false-positive rate. It uses a support vector machine trained on texture and wavelet features and includes a new algorithm to find the drop area within the image.
Iodination by VIL and HYPER-VIL.

The first high-resolution crystal structure of human D-glyceraldehyde-3-phosphate dehydrogenase is reported. The structure is used to predict possible binding sites for an anti-apoptosis compound and the E3 ubiquitin ligase Siah1.
PDB reference: GADPH, 1u8f, r1u8fsf
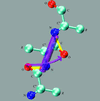
A simple and efficient method is presented to describe the secondary structure of proteins in terms of orientational distances between consecutive peptide planes and local helix parameters.

The structure of P. falciparum dihydroorotate dehydrogenase (DHODH) bound to the human DHODH inhibitor A77 1726 explains the poor binding affinity of the inhibitor and provides critical insight for the design of potential antimalarials.
PDB reference: P. falciparum dihydroorotate dehydrogenase, 1tv5, r1tv5sf

The crystal structure of the carboxy-terminal PH domain of pleckstrin is reported at 2.1 Å and comparisons are made between this structure and the recently reported solution structure of the same domain as well as other relevant PH domains.
PDB reference: pleckstrin C-terminal PH domain, 1zm0, r1zm0sf
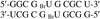
The structure of the RNA duplex reveals detailed insights of terminal and internal tandem G·U base pairs about the geometry including the stacking interactions.
PDB reference: r(GGCGBrUGCGCU)2, 2ao5, r2ao5sf
Covalent trace (<1%) labeling of proteins with a visible fluorescent probe facilitates finding crystals during the screening process.
book reviews
Free