

 journal menu
journal menu





issue contents
August 2007 issue
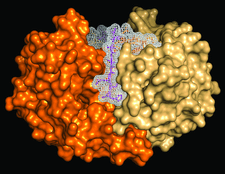
Cover illustration: B. anthracis NAD synthetase with a C-terminal His tag (magenta stick with mesh surface) bound at the interface between monomers A and B (p. 891).
research papers






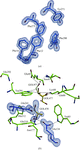
Structure–specificity relationships of an intracellular xylanase from Geobacillus stearothermophilus
The crystal structure of the intracellular xylanase (IXT6) from G. stearothermophilus T-6 has been determined and refined at 1.45 Å resolution, revealing interesting structural differences around the active site of IXT6 in comparison with the homologous extracellular xylanase (XT6) from the same bacterium.
The structure of GTB C209A from mercury-derivative and native crystals reveals partial ordering of the active-site spanning loop.

Crystal structures of unliganded wild-type and drug-resistant mutant HIV protease are reported and compared with those with bound inhibitor. A comparison of all known unliganded structures of HIV protease is made.
Cys123α of E. coli succinyl-CoA synthetase was mutated to alanine, serine, threonine and valine in order to test whether the succinyl moiety is transferred to the thiol of Cys123α as part of the catalytic mechanism. The mutant proteins were still active, but showed differences in catalysis and in structure from wild-type enzyme, demonstrating why this residue is highly conserved.
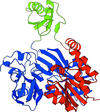
Structure of the biotin carboxylase domain of pyruvate carboxylase from Bacillus thermodenitrificans
The crystal structure of the biotin carboxylase domain of pyruvate carboxylase from B. thermodenitrificans (BC-bPC) was determined at 2.4 Å resolution. Two BC-bPC molecules are contained in the asymmetric unit to form a unique quaternary structure.

The C-terminal His tag is resolved in the structures of the apoenzyme and complexes with AMP and pyrophosphate or a substrate analog of ATP, wherein it is found to interact with native residues at the dimer interface. In the complex structures, it partially occupies the native binding site for the substrate NaAD.
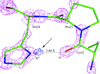
The scope of X-ray and neutron protein crystallography to determine the protonation states of functionally important amino acids is investigated at resolutions from 0.94 to 1.50 Å and from 2.2 to 3.75 Å, respectively, using concanavalin A as a test case. Key indicators are then combined to assign levels of confidence of such protonation states for three thrombin X-ray crystal structures at resolutions of 1.26, 1.32 and 1.39 Å. These various studies indicate a widening of the scope of both X-ray and neutron probes in certain circumstances to elucidate the protonation states in proteins.
short communications
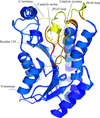
A mutation (G139A) in the MabA protein from Mycobacterium tuberculosis is reported that leads to complete protein inactivation and freezes the catalytic site into its closed form, even in the presence of the cofactor. This observation suggests a new way to develop anti-MabA drugs via protein stabilization of the `inactive' form.
PDB reference: MabA, 2ntn, r2ntnsf
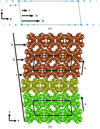
L-2-Haloacid dehalogenase from S. tokodaii strain 7 has been cloned, overexpressed and purified. The dehalogenase has been crystallized and preliminary X-ray diffraction data are reported. The crystal was shown to be an order–disorder twin and the experimental data have been detwinned.

The crystal structure of a 30 kDa protein was solved by iodide SAD using an in-house X-ray source and iodides from the crystallization liquor.
PDB reference: superoxide dismutase, 2q2l, r2q2lsf

A web server (ValLigURL; https://eds.bmc.uu.se/eds/valligurl.php) is described that can be used to compare and validate a ligand structure with reference to the structural database (wwPDB).





