journal menu
journal menu





issue contents
May 2009 issue

Cover illustration: Electrostatic interaction of the zanamivir inhibitor's functional groups with the active site of influenza A N9 neuraminidase (p. 485).
research papers






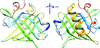
Here, the crystal structure of rat odorant-binding protein 1 is reported at 1.6 Å resolution. This protein is one of the best-characterized mammalian odorant-binding proteins and only the third such protein structure to be solved at high resolution.
PDB reference: OBP1, 3fiq, r3fiqsf
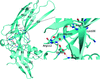
Thermodynamic and biochemical studies of the F4 fimbrial chaperone FaeE show that the dimerization observed in the crystal structure is not required for its stability in solution. Other lines of evidence indicate that self-capping of the pilin-interactive interfaces is not a mechanism that is conservedly applied by all periplasmic chaperones but rather a case-specific solution to cap aggregation-prone surfaces.
The X-ray structure of the SRP14 homologue from S. pombe has been determined using a combination of the phases obtained from anomalous scattering and radiation damage collected at a single wavelength. The combined experimental phases were of excellent quality, demonstrating that this exemplary structure from the subfamily of yeast-type Alu-domain subunits is homodimeric.
PDB reference: SRP14, 2w9j, r2w9jsf

1.8 Å X-ray crystal structure of mouse GITRL expressed in D. melanogaster S2 cells shows an identical `strand-exchanged' dimeric assembly similar to that observed previously for the E. coli-expressed protein.
PDB reference: murine GITR ligand dimer, 3fc0, r3fc0sf

The structure of BsDegV from B. subtilis was determined by sulfur-SAD phasing enhanced by bromide cocrystallization using in-house data collected using Cr Kα radiation. An innate tightly bound palmitate was found in the crystal structure.
PDB reference: BsDegV, 3fys, r3fyssf

The use of biophysical assays permitted the identification of a specific human ACC2 carboxyl transferase (CT) domain mutant that binds inhibitors and crystallizes in their presence. This mutant led to determination of the human ACC2 CT domain–CP-640186 complex crystal structure, which revealed differences in the inhibitor conformation from the yeast protein complex that are caused by differing residues in the binding pocket.
PDB reference: human ACC2 CT domain, 3ff6, r3ff6sf

A method to modify proteins with glutaraldehyde under reducing conditions is presented. Treatment with glutaraldehyde and dimethylaminoborane was found to result in cyclic pentylation of free amines and facilitated the structural determination of a protein previously recalcitrant to the formation of diffraction quality crystals.
PDB reference: EfsQnr, 2w7z, r2w7zsf
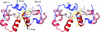
New crystal structures of the well studied protein-folding model system villin headpiece in a new space group provide insights into the conformations available in the folded state.
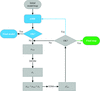
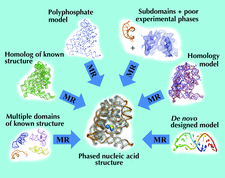
The EDM–DEDM procedure has been integrated with automated model-building programs. Correct protein models are obtained from rough phase sets derived by ab initio, SAD/MAD and molecular-replacement techniques.
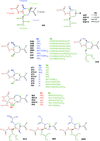
The electrostatic component of the enzyme/inhibitor interaction of a wide range influenza neuraminidases and inhibitors has been analyzed using transferable aspherical-atom densities from a recently compiled databank. Results are subdivided into the contributions of individual active-site residues and different functional groups of the inhibitors, and the effect of the Arg292→Lys mutation is considered.

Logistic regression was used to study the amino-acid composition and structure of crystal contacts in monomeric proteins. Crystal contacts are generally depleted of large flexible amino acids and enriched in small and hydrophobic residues such as Gly and Leu; additionally, larger contacts have cores depleted of polar residues.
short communications

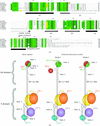
ALINE is a portable, extensible graphical sequence-alignment editor that provides a user-friendly route to editing and marking up sequence-alignment figures with maximum user control.